Helicobacter pylori
infection induit le cancer gastrique; progresser dans la recherche sur les cellules souches gastrique et les défis restant à relever
Résumé
Helicobacter pylori
infection est la principale cause de cancer de l'estomac, ce qui reste un défi de soins de santé importants. Des recherches récentes dans les cellules souches gastrique ou de la biologie des cellules progénitrices a découvert des informations précieuses pour comprendre le renouvellement de la glande gastrique et maintien de l'homéostasie, ils fournissent également des indices pour mieux définir les mécanismes par lesquels le cancer gastrique peut provenir et de progrès. LGR5, Villin-promoteur, TFF2-ARNm et Mist ont récemment été identifiés comme des marqueurs de cellules souches gastrique /progénitrices; leur identification a enrichi notre compréhension de la physiopathologie de cellules souches gastrique lors de l'inflammation chronique et la métaplasie. En outre, l'avance des marqueurs de cellules souches du cancer gastrique tels que CD44, CD90, CD133, Musashi-1 révèlent des informations inédites sur le comportement des cellules tumorales et la progression de la maladie en cause pour la thérapeutique. Cependant, deux questions essentielles restent à être des défis considérables pour l'exploration future; on est comment H. pylori
ou une inflammation chronique affecte cellules gastriques souches ou leurs progéniteurs, qui donnent lieu à mucus-, acide, pepsinogen- et lignées cellulaires hormone sécrétant. Un autre est de savoir comment une infection bactérienne ou une inflammation induit la transformation oncogénique et se propage dans les tumeurs. Focus sur les interactions de H. pylori
avec des cellules gastriques souches /progénitrices et leur microenvironnement contribuera à déchiffrer l'initiation et l'origine du cancer gastrique. Les études futures dans ces domaines seront essentiels pour découvrir les mécanismes moléculaires de la transformation oncogénique inflammation chronique à médiation et de fournir des options de prévention et d'intervention cancer. Nous passons en revue les progrès récents et de discuter des orientations futures de la recherche dans ces domaines de recherche importants.
Mots-clés
Helicobacter pylori (H. pylori)
cellules souches du cancer des cellules épithéliales gastriques épigénétique introduction
Helicobacter pylori
, un microaérophile, en forme de spirale bactérie Gram-négatif, colonisé dans l'estomac humain, est la principale cause de la gastrite chronique, des ulcères gastro-duodénaux, et des tumeurs malignes gastriques, y compris gastrique adénocarcinome non-cardia et le tissu lymphoïde muqueux associé (MALT) lymphome [1]. Épidémiologiquement, H. pylori
infecte la moitié de la population mondiale, bien que la plupart des personnes infectées ne présentent pas de présentation clinique, environ 1% des personnes infectées développeront dans le cancer gastrique [2]. Malgré la quantité suffisante des efforts pour enquêter sur ses pathogenèse et interactions hôte-pathogène, les mécanismes moléculaires de la façon dont H. pylori
infection induit le cancer gastrique restent mal compris.
Cancer gastrique (GC) est la deuxième cause de cancer lié la mort parmi tous les cancers, après le cancer du poumon. Bien que la relation entre H. pylori
infection et cancer de l'estomac a été très bien établi, les mécanismes de la façon dont l'initiation de la tumeur et le développement précoce demeurent insaisissables. Des études au cours des dernières décennies ont suggéré que les cellules souches jouent un rôle central dans l'initiation de cancers gastriques, et la tumeur peuvent provenir de cellules de la moelle osseuse dérivées [3]. Cependant, on ne sait pas comment ils donnent naissance à des tumeurs après l'insulte inflammatoire ou une infection bactérienne; dans ce travail, nous discutons brièvement les progrès actuels et des défis importants pour la recherche future.
H. pylori
facteurs de virulence et cellule hôte de signalisation dans les cellules épithéliales gastriques
Quatre principaux facteurs de virulence ont été identifiés à partir de H. pylori
(tableau 1), y compris
cytotoxine antigène associé A (CagA), cag-
îlot de pathogénicité (cag
PAI), vacuolante cytotoxine (VacA), et des protéines de la membrane externe (OMP). H. pylori cag
PAI est une région de 40 kilobases du génome de H. pylori qui code environ 30 gènes, certains d'entre eux codent quatre système de sécrétion (SSTT), qui sont essentiels pour la pathogenèse et sont responsables de la prestation de Type protéine CagA et le peptidoglycane (PGN) dans des cellules hôtes [4, 5]. CagA de H. pylori
est codée par le gène cagA de l'intérieur du PAI cag
, a un poids moléculaire d'environ 120 à 145 kDa, induit une cellule hôte multiple singaling [6]. VacA est une protéine bactérienne de 88 kDa et de la toxine, qui peut subir un clivage protéolytique limité pour donner deux sous-unités p33 et p55. Les principaux effets de VacA comprennent causant hôte vacuolisation cellulaire, l'apoptose et l'inhibition de la prolifération cellulaire [7]. OMP y compris Hop (Helicobacter Les porines de la membrane externe) et Hor (Hop-protéines apparentées) des groupes de protéines qui incluent 33 membres, leurs fonctions sont associées à une inflammation accrue gastrique muqueuse et qui sont nécessaires pour les bactéries adhésion aux cellules épithéliales [8]. L'infection par cag
PAI et H. pylori positive cagA-
souches est liée à un risque de cancer gastrique augmenté [1, 2] .Tableau 1 Major H. pylori facteurs de virulence et leurs fonctions
Nom
Taille /gènes
Effets dans la cellule hôte
référence
CagA
120-140 kDa
Oncoprotein, perturbent la polarité cellulaire, multiple signalisation
cellulaire [6]
cag
PAI
environ 30 gènes
type de système de sécrétion VI pour CagA, injection PGN et NF-kB activation
[4, 5]
88 kDa de VacA
vacuolisation, l'apoptose et l'inhibition de la prolifération cellulaire
[7]
OMP de 33 membres
inflammation et adhérence
[8]
CagA
antigène associé à la cytotoxine A; cagPAI cag
-pathogenicity île; La cytotoxine vacuolante de VacA; PGN
peptidoglycane; OMP de protéines de la membrane externe.
H. pylori
CagA et PGN sont injectées dans le cytoplasme de la cellule hôte suivant son adhésion aux cellules épithéliales gastriques, et induisent CagA- et cag
effets PAI-dépendants sur les cellules épithéliales [4, 5]. La base de la pathogenèse et la présentation clinique est que H. pylori de facteurs de virulence activent de multiples voies intracellulaires dans les cellules epitheliales, telles que les protéines kinases activées par les mitogènes (MAPK), NF-kB, la protéine d'activateur (AP) -1, Wnt /ß-caténine, transducteur de signal et activateur de la transcription 3 (STAT3) et phosphatidylinositol 3-kinase (PI3K). Leur expression conduisent à une production accrue de cytokines inflammatoires, l'infiltration de cellules immunitaires, ce qui affecte l'apoptose de la cellule hôte, la prolifération et la différenciation, finalement aboutir à la présentation clinique et les cellules épithéliales transformation oncogénique [2, 9].
Effets spécifiques CagA comprennent l'activation de Src homologie 2 contenant le domaine tyrosine phosphatase (SHP-2), en amont de Ras /Raf /Mek /Erk cascade; perturbation de la fonction épithéliale de barrière cellulaire par déphosphorylation de cortactine, ezrine, associé avec ZO-1; et la liaison au complexe Par1 /MarkII qui endommagent le contact cellulaire normale, la polarité cellulaire, et la perméabilité des cellules épithéliales [6]. CagA active également le facteur nucléaire des lymphocytes T activés (NFAT) voie de signalisation, et interagit avec la E-cadhérine de déréglementer la signalisation β-caténine, qui induit des gènes en aval β-caténine expression telle que CDX1
et favorise la cellule transdifférenciation intestinale [10 Major cag
hôte PAI-dépendante de]. signalisation implique l'introduction de PGN dans la cellule hôte cytoplasme et activer NF-kB par Nod 1 signalisation [4, 5]. L'activation de NF-kB médie l'expression de multiples gènes impliqués dans les réponses de l'hôte induites par H. pylori, tels que l'expression de l'interleukine (IL) -6, -8. En outre, des études récentes indiquent l'infection de H. pylori provoque également des changements épigénétiques dans les cellules épithéliales gastriques, y compris la méthylation de l'ADN et les modifications des histones [9]. Une enquête plus poussée dans ces domaines conduira à la découverte des mécanismes moléculaires importants dans le cancer gastrique bactéries induites.
Facteurs bactériens et hôte impliqués dans le cancer gastrique lié à l'infection par H. pylori
infection par H. pylori
chez les souris et les gerbilles, et infection de H. de la
dans les résultats de souris dans l'inflammation de l'estomac et finalement conduit à des changements histopathologiques étape-sage présentés comme la gastrite atrophique chronique, la métaplasie intestinale, la dysplasie, et adénocarcinome gastrique [11-13 ]. Les résultats impliquent des interactions synergiques entre les composants, l'inflammation et l'hôte des facteurs de Helicobacter pour la carcinogenèse gastrique. Des études récentes ont commencé à traiter des composants bactériens et hôtes individuels dans la pathogenèse.
H. pylori
CagA
H. pylori
CagA est une oncoprotéine bactérienne et est suffisante pour générer un cancer gastrique seul chez la souris. Les souris transgéniques qui surexpriment la protéine bactérienne CagA seule induisent de multiples tumeurs malignes [14], y compris l'hyperplasie épithéliale gastrique, polypes hyperplasiques, les carcinomes gastro-intestinaux et les hémopathies malignes. Les souris ne présentent aucun signe de gastrite ou d'une inflammation systémique, ainsi que les cancers sont cellules autonomes. Ces résultats indiquent directement le rôle de CagA dans la tumorigenèse gastrique, bien que les mécanismes détaillés restent à explorer plus loin.
Wnt /I²-caténine et de la cyclooxygénase 2 voies
H. pylori
infection active Wnt /β-caténine et la cyclooxygénase 2 (COX-2) /prostaglandine E (2) des voies qui jouent un rôle essentiel dans la carcinogenèse gastrique. Il est connu que l'infection par H. pylori induit β-caténine et d'expression p120 qui médient l'expression du récepteur activé par les proliférateurs des peroxysomes dans des cellules de l'épithélium gastrique et favorise gastrique prolifération des cellules épithéliales, ces effets ont été attribués à l'cag
substrats du système de sécrétion et CagA peptidoglycane [15]. Activation de la voie β-caténine conduit également à une régulation ciblée de la transcription de gènes impliqués dans la carcinogenèse, comme NFAT signalisation [10, 11]. Le plus des souris transgéniques qui surexpriment la COX2 et Wnt1 chacun seul ne produisent pas de tumeur, mais croix les deux composants pour générer des souris K19-Wnt1 /C2mE, qui expriment à la fois la COX-2 et Wnt1, provoquer le cancer chez ces souris. Dans l'estomac de ces souris, les macrophages sont également recrutés pour la muqueuse gastrique, et de promouvoir la formation de tumeurs [13, 16], ce qui suggère des actions combinées ou les effets synergiques de COX2 et Wnt les voies de signalisation sur les processus de formation de cancer.
Cytokines inflammatoires
L'infection de H. pylori
perturbe également l'homéostasie gastrique et induit de multiples production de cytokines inflammatoires au sein de la muqueuse locale, des composants tels que l'IL-1β, le TNF-α et IL-10 génotypes sont associés à un risque accru de développer un cancer gastrique [ ,,,0],17-19] de. IL-1β est une cytokine pro-inflammatoire impliquée dans l'inflammation et l'immunité. polymorphismes IL-1 sont associés à une production accrue d'IL-1β et un risque accru de cancer gastrique [19], l'IL-1β inhibe également la sécrétion d'acide gastrique. Chez les souris transgéniques, de l'estomac surexpression spécifique IL-1β induit l'inflammation gastrique, métaplasie, la dysplasie et le carcinome progressive spontanée.
L'infection de H. dans une progression plus rapide à l'atrophie gastrique et le cancer. La surexpression de l'IL-1β mobilise également les cellules suppressives dérivées myéloïdes et induit l'activation de NF-kB, ainsi que ses gènes en aval de l'IL-6, l'expression de TNF-α dans ces cellules. En outre, l'IL-1β seule est suffisante pour induire une pré-néoplasie gastrique [17]; Cependant, on ne sait pas les mécanismes de la façon dont l'IL-1β sur l'expression se traduit finalement dans la transformation oncogénique à l'heure actuelle. Le plus intéressant, d'autres médiateurs inflammatoires peuvent exercer des effets opposés. Un exemple est l'IFN-γ, qui est produit principalement par les lymphocytes T activés, les cellules tueuses naturelles et est un médiateur clé de l'immunité innée et adaptative. IFN-γ médie les réponses à une infection bactérienne et une maladie auto-immune, et agit en tant que suppresseur de tumeurs [18]. Chez la souris, de l'estomac surexpression spécifique de l'IFN-γ seul a des effets minimaux sur la muqueuse gastrique, mais inhibe l'IL-1β- et H. gastrite et de la néoplasie induite par
. Le mécanisme a été attribué à l'IFN-γ inhibe gastrique prolifération des cellules épithéliales, accélère l'apoptose des lymphocytes T gastriques et diminue la production de cytokines pro-inflammatoires Th1 et Th17. Ces effets peuvent équilibrer la prolifération des cellules épithéliales, empêcher l'inflammation, et finalement inhiber la formation de tumeurs [18]. Les facteurs minette de Par conséquent, la perturbation de la cellule hôte inflammatoire production de cytokines participe dans l'oncogenèse gastrique.
Les protéines de la famille des facteurs de lotier (TFF1, 2, 3), réguler la réparation des muqueuses et réprimer la formation de tumeur dans l'estomac. TFF1 et TFF2 ont été récemment identifiés à jouer un rôle critique dans antagoniser carcinogenèse gastrique [20, 21]. TFF1 de ses actes comme un gène suppresseur de tumeur et de ses résultats de carence dans le cancer gastrique spontanée chez la souris [21]. TFF2
souris déficientes seulement afficher des altérations subtiles dans la muqueuse prolifération; activation des cellules pariétales présenté comme une sécrétion accrue d'acide et une sensibilité accrue aux blessures AINS. L'ablation génétique de TFF2
accélère la croissance tumorale et favorise les lésions précancéreuses chez les souris antrale [20]. Chez ces souris, un déséquilibre de l'hormone et la production de cytokines a été notée, avec une diminution gastrokine 1, 2 la production d'ARNm et a augmenté la réponse Th1, une diminution de la réponse Th2 (IL-1α, β et l'interféron-γ, une réduction de l'IL-13, IL-4) [ ,,,0],20, 21].
deux TFF1 et l'expression de TFF2 sont souvent perdus dans le cancer gastrique et aberrante méthylation du promoteur a été suggéré d'être un mécanisme important. Il est intéressant de
infection par H. augmente la perte de TFF1 et H. pylori
(SS1 souche) une infection augmente le promoteur de la méthylation de TFF2 chez les souris qui se traduit dans le niveau inférieur de l'expression des protéines [20, 21] . . Par conséquent, ces données révèlent des effets protecteurs des protéines de la FFT et leur réduction promouvoir cancérogenèse gastrique
Ensemble, les systèmes ci-dessus ont clairement fait progresser notre compréhension du rôle de H. pylori
hôtes ou facteurs dans la carcinogenèse gastrique; bien que les données sont assez fragmentaires, ils peuvent être convergents et potentiellement un impact sur les cellules souches ou progénitrices gastriques vers cancérogenèse. En effet, le rôle des cellules souches ou progénitrices dans ces systèmes modèles a juste commencé à être apprécié. Des études ont cherché à remédier l'initiation de la tumeur et la transformation maligne en se concentrant sur les cellules souches, microenvironnement, et le recrutement de diverses cellules progénitrices qui contribuent à la croissance de la tumeur, ce qui sera discuté plus en détail ci-dessous. Le plus des cellules souches ou progénitrices gastriques et les cellules épithéliales gastriques de leurs marqueurs qui constituent le presse-étoupe dans la muqueuse gastrique sont creux, pariétal, le cou et les cellules zymogènes, ainsi que des cellules progénitrices. Deux limites importantes dans la recherche sur les cellules souches sont le manque d'in vitro
système de culture et le manque de marqueurs spécifiques pour les différentes étapes de cellules souches. Des études récentes commencent à identifier ces cellules en traçant l'analyse génétique de la souris de la lignée, qui est un outil puissant pour la caractérisation et la validation des marqueurs de cellules souches et leurs fonctions.
Le concept original de l'estomac hypothèse sur les cellules souches du cancer est que les cellules situées dans l'isthme la région de la glande gastrique, la cellule immature morphologiquement moins organoïde, sont des cellules souches multipotentes; ils sont responsables de la génération des quatre principaux types de cellules de la glande gastrique [22]. Ces cellules souches donnent lieu à transit amplification (/progénitrices de cellules filles), qui peut se différencier en tous les types de cellules matures. Au cours de ce processus, les descendants issus de cellules souches subissent une migration bipolaire complexe de la région cou /isthme, se déplaçant vers le haut ou vers le bas. Dans l'estomac de la souris, les trois types de cellules progénitrices (précuvettes, preneck, et les cellules preparietal) ont été trouvés provenir de cellules libres granulaires-multipotentes dans isthme région [22]. Toutefois, ce modèle a récemment été enrichi avec la découverte de la prolifération Villin-promoteur- et les cellules (voir ci-dessous) à la base et les différents emplacements des glandes pyloriques, qui présentent à la fois auto-renouvellement et la capacité de générer tous les épithéliales différenciées LGR5-marqué les types de cellules de la glande pylorique [23, 24]. Ces observations ont ouvert de nouvelles voies pour étudier gastrique la biologie des cellules souches et pathobiologie.
Lignage traçage génétique marquée des cellules souches gastriques
Plusieurs souches ou cellules progénitrices marqueurs gastriques récemment identifiés à l'aide de lignage génétique traçage technologie sont répertoriés dans le tableau 2.Table 2 souches et cellules progénitrices marqueurs gastriques
Nom
Lieu
Référence
LGR5
Antral
Fonction
, base de la glande
Susciter à l'unité gastrique, les quatre types de cellules
[23]
Villin-promoteur
Antral, base de la glande
donner lieu à l'unité gastrique, les quatre types de cellules
[24]
TFF2 ARNm
Corpus, la base glande
donner lieu à seulement pariétal, le cou et les cellules principales
[27]
Mist1 (BHLHA15)
Corpus, la base glande
cellule en chef de génération de SPEM
[28]
leucine-riche contenant répétées couplé aux protéines G de receptor5 de LGR5 (LGR5); Villin-promoteur Villin-promoteur marqué les cellules souches; TFF2 ARNm
, facteur de Trifold 2 ARNm marqué des cellules souches; Mist1 (BHLHA15)
, base familiale hélice-boucle-hélice, membre a15 de. LGR5 marqué les cellules souches gastriques
Utilisation in vivo
lignage analyse de traçage, Barker et ses collègues [23] démontrent que LGR5 ( les cellules riches en leucine G récepteur couplé aux protéines contenant repeat-5, GPR49) marquées sont longues en direct; résider dans la base de la glande, morphologiquement immature, avec un grand rapport et limitées organites nucléaires à-cytoplasmique. LGR5 est un gène cible identifié Wnt dans des lignées cellulaires de cancer du côlon et cryptes intestinales. Il est récemment identifié comme un nouveau marqueur de cellules souches de l'estomac, de l'épithélium intestinal et du follicule pileux [23].
Dans néonatale souris estomac [23], LGR5 est exprimée à la base du corpus et pyloriques glandes, alors que chez les souris adultes LGR5 est essentiellement limité à la base des glandes pyloriques matures. Contrairement à la notion commune que les cellules souches sont au repos, LGR5 marqué les cellules, prolifèrent rapidement et sont en mesure de construire la totalité de la glande gastrique sur une courte période. Au cours de la culture in vitro
, cellule unique positif LGR5 généré efficacement organites ressemblant à long terme épithélium pylore mature avec capacité d'auto-renouvellement, l'architecture et la composition cellulaire.
Étude du transcriptome des cellules LGR5 [23] révèlent total de 153 changé l'expression des gènes dans LGR5 souches population cellulaire cellulaire /fille, beaucoup sont des gènes cibles de Wnt, y compris CD44, Sox9, Sord, Prss23, Sp5, indiquant une activité de signalisation Wnt canonique à la base des glandes pyloriques. Plusieurs gènes spécifiques entéro, y compris Chromogranin A &. LGR5 souches les cellules B, somatostatine et Gastrin G, sont fortement régulés positivement dans la population LGR5 de cellule fille, ce qui implique une différenciation rapide vers la lignée entéroendocrine sont également tumorigène, les souris avec suppression d'APC gène, un gène essentiel de la signalisation Wnt a entraîné une régulation positive β-caténine à la base des glandes pyloriques et dans les 2-3 semaines, ces cellules peuvent se développer dans LGR5 adénomes hautement prolifératifs, β-caténine positif. Cet effet n'a pas été détecté dans corpus gastrique, confirmant l'absence de LGR5 dans cette région [23].
En raison de ces découvertes passionnantes, la distribution de cellules LGR5 est examinée plus avant dans l'estomac humain. Immunocoloration a révélé la délocalisation des cellules LGR5 à différents stades de maladies gastriques [25]. En non-néoplasique muqueuse de l'estomac, les cellules LGR5 se trouvent principalement dans la région du cou muqueuse; lors de la métaplasie intestinale, ils se localisent à la base crypte; et GC, les cellules LGR5 sont présents au, centre de la tumeur surface luminale et le front d'invasion. Répartition des cellules LGR5 dans le centre de la tumeur et l'invasion avant du GC est bien corrélée avec la croissance de la tumeur et la propagation nodal. En outre, les patients atteints de LGR5 positifs glucocorticoïdes ont une médiane de survie plus courte que les patients atteints de LGR5 GCS négatifs. les tissus tumoraux provenant de l'œsophage, de l'estomac, du foie, du pancréas, du côlon et du rectum montrent tous significativement plus de cellules LGR5 et des niveaux plus élevés d'expression de l'ARNm-LGR5 par rapport aux tissus non tumorales [25].
Ensemble, ces observations chez la souris et l'homme ont documentés que les cellules souches sont beaucoup plus mobiles et moins restreint aux positions comme ils étaient à l'origine des pensées. En outre, LGR5 marqué les cellules cancéreuses peuvent proliférer vers différentes directions à l'intérieur de la muqueuse, ce qui indique une propriété de croissance invasive et implique qu'elles sont cruciales pour le développement du cancer de l'estomac et de la progression. D'autres études sont nécessaires pour comprendre leur rôle dans la tumorigenèse gastrique surtout pendant le réglage de l'infection par H. pylori chronique. Fait intéressant, la tendance a déjà commencé; rapport pilote a montré que H. pylori
infection est associée à une augmentation des dommages de l'ADN de cellules positives LGR5 chez les patients atteints de cancer gastrique [26].
Villin-promoteur marqué les cellules souches gastriques
Villin est un actine-Attaches- des protéines, principalement exprimée dans la bordure en brosse de l'épithélium intestinal, joue un rôle clé dans la morphogenèse des microvillosités pour former des protubérances de la membrane cellulaire qui augmentent la surface et qui impliquent l'absorption cellulaire, la sécrétion et l'adhérence. Villin est fortement exprimé dans la cellule intestinale et largement dépourvue de cellules épithéliales gastriques.
Lineage traçage études confirment que les cellules marquées Villin-promoteur peuvent être trouvés dans les souris épithélium gastrique, ils sont au repos et de localiser au niveau ou en dessous de l'isthme dans le tiers inférieur une partie des glandes pyloriques et rare dans corpus [24]. Ces cellules donnent naissance à de multiples lignées gastriques des glandes antrales, y compris la cellule de la fosse de surface (cellules des glandes muqueuses), des cellules entéro-endocrines, et les cellules pariétales. Cependant, endogène expression de la protéine villine est pas observée dans l'épithélium gastrique de ces souris probablement en raison de perturbations génétiques structurelles; il est donc difficile d'établir la relation précise des populations de cellules souches actives et inactives. Semblable à LGR5, Villin-promoteur marqué souches population de cellules sont également absents dans la région de corpus et d'indiquer différentes biologie des cellules souches dans cette région de l'estomac [24].
Une conclusion importante de ce travail élégant est que l'exposition de l'IFN-γ forcé chez la souris pour imiter cause l'inflammation remarquable expansion du Villin-promoteur marqué population de cellules dans la muqueuse antrale [24]. Les cellules marquées peuvent être trouvées pour localiser à différentes positions dans la partie inférieure de la glande, entre isthme et la pointe de la glande, et situé sur le côté opposé de la base des glandes, ce qui suggère une inflammation régule ces prolifération cellulaire et la migration. Tout en utilisant des souris transgéniques CDX2, un modèle pour induire la métaplasie intestinale, trouver aucun changement significatif dans les deux nombre de cellules et emplacements, ce qui implique des cellules marquées Villin-promoteur ne donnent pas lieu à la métaplasie. Il serait intéressant de voir si d'autres médiateurs inflammatoires pourraient avoir un impact sur ces cellules et modifier leur amplification et la mobilité ainsi
facteur Trefoil 2 ARNm marqué tige gastrique /cellules progénitrices
Dans la glande gastrique., Les cellules du col de mucus localiser en dessous de la région de l'isthme et de la famille du facteur de lotier express 2 protéines; mais transcrits d'ARNm TFF2 sont concentrées dans les cellules au-dessus de la région de col dans la muqueuse normale du corpus. Ce changement de position suggère que TFF2 ARNm des cellules de transcription exprimant (TTE) est peut-être des cellules progénitrices gastriques et les transcriptions TFF2 ARNm peut être un marqueur [27].
Dans la muqueuse oxyntique [27], de la lignée de traçage démontrent que TFF2 ARNm marqué cellules migrent vers le bas de la glande, donner lieu à pariétal, le cou muqueuse, et le chef (zymogène) lignées cellulaires, mais pas entérochromaffines-like-cellule. cellules de mucus de surface ne sont pas dérivées de cellules TTE et la descendance de la lignée TTE ne survivent pas au-delà de 200 jours [27]. Contrairement au corpus dans l'antre, les cellules TTE ne semblent pas migrer, ni ils représentent tous les types de cellules souches ou progénitrices dans l'estomac distal, suggérant TTE a marqué les cellules uniquement situés dans le corpus gastrique. Toutefois, les données fonctionnelles ne sont pas disponibles à partir de cette étude, les futures études sont nécessaires pour comprendre son rôle dans le renouvellement de la glande gastrique et la différenciation lors de l'inflammation.
Mist1
marqués gastriques cellules souches /progénitrices
Mist1 (BHLHA15) est un facteur de transcription, exprimé dans les cellules principales et est critique pour le chef localisation cellulaire basale et la maintenance. Elle est exprimée à la base de la glande gastrique et dans les cellules situées dans les zones de transition entre le cou et la région de base avec les caractéristiques des cellules du col chef partielle [28].
Lineage traçage dans des modèles de souris atrophie oxyntiques aiguës et chroniques avec des cellules Mist1 exprimant (traitement à l'aide chimique L-635 et
infection H.) démontrent que cellule principale peut donner lieu à l'ensemble de la métaplasie (SPEM) lignée polypeptide exprimant spasmolytique. SPEM est une forme récemment apprécié de l'estomac métaplasie de la muqueuse, en plus de la métaplasie intestinale, SPEM get it nom en raison de elle exprime TFF2 (polypeptide spasmolytique), également appelé métaplasie pseudopyloric ou métaplasie muqueuse ou antralization du corpus [29]. les cellules en chef peuvent donc représenter des cellules progénitrices pour métaplasie [28]. La perte de cellules pariétales induit la transdifférenciation des cellules en chef SPEM et ce métaplasie peut subir une expansion sous l'influence de l'inflammation aiguë ou chronique.
Atrophie gastrique du corps et le corps sont associés au développement de SPEM et SPEM est fortement associée à la le développement du cancer gastrique similaire à la métaplasie intestinale. Les deux SPEM et métaplasie intestinale sont observées dans l'estomac des patients atteints de cancer gastrique. SPEM possède les caractéristiques de métaplasie antrale et expriment TFF2 et MUC6, tandis que la métaplasie intestinale présente des caractéristiques de la lignée claires de duodénum de l'intestin, avec l'expression de TFF3 et MUC2 [28, 29]. À l'heure actuelle, on ne sait pas si le cancer gastrique pourrait provenir de l'un ou aucun des deux types de métaplasie, les travaux futurs sont garantis pour évaluer leur relation et le rôle de SPEM dans la carcinogenèse gastrique.
Putatifs cellules souches gastrique et souches du cancer marqueurs cellulaires
Pour résumé, quelques-uns ont récemment rapporté des marqueurs de cellules de cellules souches et de la tige de cancer gastrique putatifs sont énumérés dans le tableau 3.Table 3 putatifs cellules souches gastrique et souches cancéreuses des marqueurs de cellules
Nom
Lieu
autorenouvellement
xénogreffe
Référence
CD44
base de la glande
formation
sphère tumorigène
[32]
CD90
n /t
formation sphère
tumorigènes [35]
CD133
base de la glande
oui
tumorigènes [38]
Musashi-1
antrale, isthme /cou
n /t
n /t
[41]
pSmad2 /3L-Thr
région isthme
n /t
n /t
[43]
CD de cluster de différenciation; n /t
non testé; pSmad2 /3L-Thr
Grappe de Smad2 /3 thréonine phosphorylée au niveau de liaison spécifique. différenciation 44 (CD44)
CD44 est une glycoprotéine transmembranaire de la surface cellulaire qui agit comme un récepteur pour les matrices extracellulaires tels que l'acide hyaluronique, sa fonction consiste dans l'interaction cellule-cellule, l'adhésion cellulaire et la migration. CD44 est une cible en aval connu de la voie Wnt /β-caténine et est exprimée dans de nombreux types de tissus et de tumeurs solides telles que l'estomac, le pancréas, les cancers du côlon [30-33]. CD44 a une variété de variants d'épissage (CD44v) [30] et est identifiée comme étant une souche tumorale solide marqueur de cellules du cancer du sein [31]. cellules CD44 exprimant ont le cancer cellules souches (CSC) dispose notamment; tumorigène, capable d'auto-renouvellement et générer des phénotypique diverses (populations mixtes de) cellules non tumorigènes.
Dans SCID (sévère de déficit immunitaire combiné) des souris, les cellules cancéreuses gastriques positives CD44 sont capables d'induire une tumeur [32], CD44 knockdown en court en épingle à cheveux l'ARN diminue la tumorigénicité. En outre, ces cellules sont résistantes à la chimiothérapie ou la radiothérapie. Dans la même expérience, d'autres marqueurs du SCC potentiels, tels que CD24, CD133, CD166, spécifique au stade embryonnaire antigène-1 (SSEA-1), et SSEA-4, ou le tri pour la population de côté de cellules ne présentent aucune corrélation avec la tumorigénicité
in vitro ou in vivo
[32]. En outre, de récents rapports indiquent CD44, ADAM17 et un autre marqueur putative de cellules souches, Musashi-1 coexprime avec des marqueurs de cellules souches LGR5 dans les deux tissus de patients cancéreux normaux et gastriques [25]. Es expression de CD44 variantes chez les patients atteints de cancer gastrique tels que CD44v6 et CD44v9 sont associés à la progression du cancer gastrique [30]. Expression de CD44v6 est également associée à des métastases ganglionnaires, l'invasion et le grade pathologique de la tumeur à un stade avancé, les patients atteints de tumeurs positives CD44v6 ont un taux de survie plus faible [34]. Cd44v9 est pas exprimé sur l'épithélium gastrique de H. pylori de l'individu négatif, mais est présent lors de H. pylori
infection [30]. Toutefois, si les variantes de CD44 peuvent servir de fabricants de cellules souches du cancer est en attente de nouvelles investigations.
Cluster de différenciation 90 (CD90)
CD90 (Thy-1) est une glycoprotéine de surface cellulaire, le poids moléculaire à 25-37 kDa . Chez les souris, il est exprimé dans des thymocytes, des neurones, des cellules T; chez l'homme, CD90 est exprimé dans l'endothélium, les muscles lisses, des fibroblastes et une variété de cellules souches; y compris la moelle osseuse des cellules dérivées de souches mésenchymateuses, la tige hépatique /cellules progénitrices, des cellules souches hématopoïétiques et les cellules souches kératinocytaires [35, 36]. CD90 marqué les cellules souches cancéreuses ont été identifiées à partir du cancer du foie humain, la tumeur murine du sein et dans les échantillons stroma de tumeurs gastro-intestinales cliniques [36, 37].
CD90 a été récemment rapporté pour caractériser la population SCC dans les tumeurs primaires gastriques [35]. Dans les cellules humaines primaires isolées de tumeurs gastriques, CD90 agit comme un marqueur de surface et peuvent être enrichies dans des conditions non-adhérentes, sans sérum et les conditions de sphère filmogène.
 Faire face à la maladie cœliaque
Faire face à la maladie cœliaque
 Les enfants sont immunisés contre le SRAS-CoV-2
Les enfants sont immunisés contre le SRAS-CoV-2
 Symposium scientifique à LABVOLUTION se concentre sur les questions clés des sciences de la vie
Symposium scientifique à LABVOLUTION se concentre sur les questions clés des sciences de la vie
 Les bactériophages pourraient traiter E. coli sans endommager l'intestin,
Les bactériophages pourraient traiter E. coli sans endommager l'intestin,
 Vas-y,
Vas-y,
 Transmission mère-enfant du SRAS-CoV-2 pendant la grossesse possible mais rare,
Transmission mère-enfant du SRAS-CoV-2 pendant la grossesse possible mais rare,
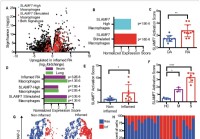 Un nouveau récepteur de macrophages super-activant pourrait expliquer l'hyper-inflammation dans les cas graves de COVID-19
Limmunité est une chose curieuse. Bien quessentiel pour protéger le corps contre les agents pathogènes envahissants et les antigènes étrangers, il peut également se retourner contre le corps et déclen
Un nouveau récepteur de macrophages super-activant pourrait expliquer l'hyper-inflammation dans les cas graves de COVID-19
Limmunité est une chose curieuse. Bien quessentiel pour protéger le corps contre les agents pathogènes envahissants et les antigènes étrangers, il peut également se retourner contre le corps et déclen
 Une approche multi-omique pour le développement de médicaments contre le COVID-19
Une nouvelle étude publiée sur le serveur de préimpression medRxiv* en mai 2020 rapporte une approche multi-omique qui pourrait faciliter le développement de médicaments efficaces contre COVID-19. L
Une approche multi-omique pour le développement de médicaments contre le COVID-19
Une nouvelle étude publiée sur le serveur de préimpression medRxiv* en mai 2020 rapporte une approche multi-omique qui pourrait faciliter le développement de médicaments efficaces contre COVID-19. L
 E. coli superbactérie se propageant par une mauvaise hygiène des toilettes,
pas par la nourriture Une nouvelle étude publiée dans Les maladies infectieuses du Lancet le 22 octobre 2019, dit quun superbactérie commun qui cause plus de 5, 000 cas dintoxication alimentaire en
E. coli superbactérie se propageant par une mauvaise hygiène des toilettes,
pas par la nourriture Une nouvelle étude publiée dans Les maladies infectieuses du Lancet le 22 octobre 2019, dit quun superbactérie commun qui cause plus de 5, 000 cas dintoxication alimentaire en