 Karzinoide können krebsartig oder gutartig sein und bilden sich hauptsächlich in Lunge und Darm. Der klinische Verlauf dieser Krebsarten ist oft träge mit einer 5-Jahres-Überlebensrate von annähernd 75 %.
Karzinoide können krebsartig oder gutartig sein und bilden sich hauptsächlich in Lunge und Darm. Der klinische Verlauf dieser Krebsarten ist oft träge mit einer 5-Jahres-Überlebensrate von annähernd 75 %.
Hitzewallungen oder Hitzewallungen gelten als charakteristisches Symptom des Übergangs in die Wechseljahre; Sie können jedoch auch bei Männern aufgrund von Zuständen wie niedrigem Testosteron auftreten. Flush kann bei Menschen auch als Folge bestimmter seltener Erkrankungen wie dem Karzinoid-Syndrom auftreten, einer Erkrankung, bei der ein Karzinoid-Tumor große Mengen an Serotonin, einem vom Körper produzierten Hormon, absondert.
Weitere Symptome des Karzinoidsyndroms oder Karzinoidtumors sind:
Ein karzinoider Tumor ist ein Tumor, der sich aus enterochromaffinen Zellen entwickelt. Enterochromaffine Zellen sind hormon- und chemikalienproduzierende Zellen, die normalerweise im Dünndarm, Blinddarm, Dickdarm, Rektum, Bronchien, Bauchspeicheldrüse, Eierstöcken, Hoden, Gallengängen, Leber sowie anderen Organen zu finden sind. Enterochromaffine Zellen produzieren viele Arten von Substanzen, zum Beispiel Histamin, Serotonin, Dopamin, Tachykinine und andere Chemikalien, die tiefgreifende Auswirkungen auf das Kreislaufsystem (Herz und Blutgefäße), den Magen-Darm-Trakt und die Lunge haben. Diese Substanzen werden daher als vasoaktive Amine bezeichnet. Beispielsweise kann Serotonin aufgrund der Erweiterung der Blutgefäße Durchfall, Histamin-Keuchatmung und Tachykinin-Flush verursachen.
Da sich karzinoide Tumoren aus enterochromaffinen Zellen entwickeln, behalten sie häufig die Fähigkeit, dieselben Hormone zu produzieren, oft in großen Mengen. Wenn diese Hormone im Blut zirkulieren, können sie Symptome des Karzinoidsyndroms verursachen, das später besprochen wird.
Das wichtige Merkmal von Karzinoidtumoren, das sie von anderen Tumoren des Gastrointestinaltrakts unterscheidet, ist ihr Potenzial, das Karzinoidsyndrom zu verursachen. Die meisten anderen Tumoren des Magen-Darm-Trakts (wie Dickdarmkrebs oder Dünndarm-Lymphom) verursachen hauptsächlich aufgrund ihrer lokalen Auswirkungen auf den Darm Symptome wie Bauchschmerzen, Darmblutungen und Darmverschluss. Karzinoidtumoren können zwar auch diese lokalen Symptome verursachen, sie können aber auch die Substanzen produzieren und freisetzen, die das Karzinoidsyndrom verursachen. Oft können die Symptome des Karzinoidsyndroms verheerender sein als die lokalen Symptome.
Das Karzinoid-Syndrom ist eine Kombination von Symptomen, die durch Hormone und andere chemische Substanzen verursacht werden, die von den Tumoren in den Blutkreislauf abgegeben werden. Die Symptome des Karzinoid-Syndroms variieren je nachdem, welche Hormone von den Tumoren ausgeschüttet werden. Die üblicherweise freigesetzten Hormone sind Serotonin, Bradykinin (ein Molekül, das von Enzymen an der Stelle einer Verletzung produziert wird und dann an Rezeptoren bindet, um Schmerzen zu verursachen), Histamin und Chromogranin A (ein allgemeiner Marker für neuroendokrine Tumore).
6 Zu den typischen Karzinoidsymptomen gehören:
Herzerkrankungen treten bei geschätzten 50 % der Patienten mit dem Karzinoid-Syndrom auf. Das Karzinoidsyndrom verursacht typischerweise Narbenbildung und Steifheit der Trikuspidal- und Pulmonalklappen der rechten Herzseite. Die Steifheit dieser beiden Klappen verringert die Fähigkeit des Herzens, Blut aus der rechten Herzkammer in die Lunge und den Rest des Körpers zu pumpen, und führt zu Herzversagen.
Typische Symptome einer Herzinsuffizienz sind ein
Die Schädigung der Trikuspidal- und Pulmonalklappen des Herzens beim Karzinoidsyndrom wird höchstwahrscheinlich durch eine längere Exposition gegenüber hohen Serotoninspiegeln im Blut verursacht.
Die Karzinoidkrise ist ein gefährlicher Zustand, der zum Zeitpunkt einer Operation auftreten kann. Es ist gekennzeichnet durch einen plötzlichen und starken Abfall des Blutdrucks, der einen Schock verursacht, manchmal begleitet von einer ungewöhnlich schnellen Herzfrequenz, hohem Blutzucker und schwerem Bronchospasmus. Eine Karzinoidkrise kann tödlich sein. Der beste Weg zur Vorbeugung einer Karzinoidkrise ist die Behandlung von Patienten, die sich einer Operation unterziehen, mit Somatostatin (siehe unten), bevor die Operation beginnt.
Keuchen tritt bei etwa 10 % der Patienten mit Karzinoid-Syndrom auf. Es ist das Ergebnis eines Bronchospasmus (Verengung der bronchialen Atemwege), der durch Hormone verursacht wird, die von den karzinoiden Tumoren freigesetzt werden.
Bauchschmerzen sind bei Patienten mit Karzinoidsyndrom häufig. Die Schmerzen können auf Tumormetastasen in der Leber, Tumorinvasion benachbarter Gewebe und Organe oder einen tumorverursachenden Darmverschluss zurückzuführen sein (siehe Dünndarm-Karzinoidtumoren unten).
Die Prävalenz karzinoider Tumore ist schwer zu bestimmen, da viele karzinoide Tumore nicht erkannt werden, weil sie keine Symptome hervorrufen. In einer Autopsieserie wurde geschätzt, dass karzinoide Tumoren eine Prävalenz von 8/100.000 Menschen pro Jahr haben, aber 90 % der bei der Autopsie gefundenen Karzinoide waren nicht die Todesursache.
Das Karzinoid-Syndrom ist sehr selten. Dies liegt daran, dass viele karzinoide Tumoren nicht sekretorisch sind – das heißt, sie produzieren nicht die Hormone, die für das Karzinoid-Syndrom verantwortlich sind. Ein weiterer Grund für die Seltenheit des Syndroms ist, dass die Chemikalien, die von karzinoiden Tumoren freigesetzt werden, insbesondere von Tumoren im Unterleib, oft von der Leber zerstört werden, bevor sie den allgemeinen Kreislauf erreichen und Symptome verursachen. Beispielsweise werden die von Karzinoidtumoren des Dünndarms produzierten Chemikalien in die Pfortader freigesetzt. Das Pfortaderblut passiert die Leber, bevor es das Herz und den allgemeinen Kreislauf erreicht. Wenn das Pfortaderblut durch die Leber fließt, werden die Hormone von der Leber inaktiviert oder zerstört.
Nur solche Karzinoidtumoren, die diese Substanzen direkt in den allgemeinen Kreislauf abgeben können (und nicht in die durch die Leber verlaufenden Pfortadern), können das Karzinoidsyndrom hervorrufen. Die häufigste Ursache des Karzinoidsyndroms sind daher Dünndarm-Karzinoidtumoren, die in die Leber metastasiert haben. Die Metastasen in der Leber können die Chemikalien direkt in den Kreislauf abgeben. Ein weiteres seltenes Beispiel sind karzinoide Tumoren der bronchialen Atemwege. Karzinoide Tumore in den bronchialen Atemwegen können ihre vasoaktiven Amine direkt über die Lungenvenen in den allgemeinen Kreislauf abgeben, ohne die Leber zu passieren.
Karzinoide Tumoren können überall dort gefunden werden, wo es enterochromaffine Zellen gibt, im Wesentlichen im ganzen Körper. Die Mehrzahl (65 %) der Karzinoidtumoren findet sich im Gastrointestinaltrakt. Der Ursprung von gastrointestinalen Karzinoidtumoren ist am häufigsten der Dünndarm, Blinddarm und Rektum. Weniger häufige Ursprünge sind Magen und Dickdarm; und die am wenigsten verbreiteten Ursprünge sind Bauchspeicheldrüse, Gallenblase und Leber (obwohl karzinoide Tumore in der Leber normalerweise Metastasen von anderswo sind).
Etwa 25 % der karzinoiden Tumoren finden sich in den bronchialen Atemwegen und der Lunge. Die restlichen 10 % sind fast überall zu finden. In manchen Fällen können Ärzte den Ursprungsort der Karzinoidtumoren nicht lokalisieren, obwohl sie anhand der Symptome des Karzinoidsyndroms wissen, dass sie vorhanden sind.
Karzinoide des Dünndarms
Im Allgemeinen sind Dünndarmtumoren (ob gutartig oder bösartig, Adenokarzinome oder Karzinoide) selten und viel seltener als Dickdarm- oder Magenkrebs. Dennoch machen Karzinoidtumoren ein Drittel aller Dünndarmtumoren aus und werden am häufigsten im Ileum (dem unteren Teil des Dünndarms in der Nähe des Dickdarms) gefunden. Karzinoide Dünndarmtumoren verursachen typischerweise keine Symptome oder erzeugen nur vage Bauchschmerzen. Daher ist es schwierig, Karzinoidtumoren des Dünndarms frühzeitig zu erkennen, während sie noch vollständig entfernt und der Patient geheilt werden können. Die wenigen kleinen Karzinoide, die früh entdeckt werden, werden meist zufällig bei Röntgenaufnahmen oder Eingriffen zu anderen Zwecken entdeckt. Typischerweise werden Dünndarmkarzinoide erst spät diagnostiziert, oft Jahre nach Symptombeginn und meist nachdem bereits lokale und entfernte Metastasen vorhanden sind.
Etwa 10 % der Karzinoide des Dünndarms verursachen ein Karzinoidsyndrom. Das Vorliegen eines Karzinoidsyndroms bedeutet normalerweise, dass der Tumor bösartig ist und sich auf die Leber ausgebreitet hat.
Dünndarmkarzinoide verstopfen oft den Dünndarm, wenn sie eine große Größe erreichen. Zu den Symptomen eines Dünndarmverschlusses gehören krampfartige Bauchschmerzen, Übelkeit und Erbrechen und manchmal Durchfall. Obstruktion kann durch zwei verschiedene Mechanismen verursacht werden. Der erste Mechanismus ist die Vergrößerung und das Wachstum des Tumors in das Lumen (Kanal) innerhalb des Dünndarms. Der zweite Mechanismus ist ein Abknicken des Dünndarms aufgrund einer fibrosierenden Mesenteritis, einem Zustand, der durch den Tumor verursacht wird, bei dem eine ausgedehnte Narbenbildung in dem den Dünndarm umgebenden Gewebe auftritt. Fibrosierende Mesenteritis verstopft manchmal die Arterien, die den Darm mit Blut versorgen, was zum Absterben eines Teils des Darms führt (Gangrän). Der gangränöse Darm kann platzen und lebensbedrohlich sein.
Während Tumoren des Blinddarms selten sind, sind karzinoide Tumoren der häufigste Tumor des Blinddarms und machen etwa die Hälfte aller Blinddarmtumoren aus. Tatsächlich werden Karzinoide in 0,3 % der resezierten (entfernten) Anhänge gefunden, aber die meisten von ihnen sind kleiner als 1 cm und verursachen keine Symptome. Sie befinden sich hauptsächlich in Anhängen, die aus anderen Gründen entfernt wurden. Die meisten Behörden sind der Ansicht, dass die Appendektomie eine angemessene Behandlung für diese kleinen karzinoiden Tumoren des Blinddarms ist. Die Wahrscheinlichkeit, dass ein Tumor nach einer Appendektomie erneut auftritt, ist sehr gering. Appendixkarzinoidtumoren, die zum Zeitpunkt der Diagnose größer als 2 cm sind, haben eine Wahrscheinlichkeit von etwa 30 %, bösartig zu sein und lokale Metastasen zu haben. Daher erfordern größere karzinoide Tumoren des Anhangs eine umfangreichere Operation, wie z. B. die Entfernung des rechten Dickdarms, anstelle einer einfachen Appendektomie. Große Appendixkarzinoide sind glücklicherweise selten. Karzinoidtumoren, die auf den Blinddarm beschränkt sind, selbst Metastasen in lokalen Geweben, verursachen normalerweise kein Karzinoidsyndrom.
Rektumkarzinome werden oft zufällig bei einer flexiblen Sigmoidoskopie oder Koloskopie entdeckt. Das Karzinoidsyndrom ist bei rektalen Karzinoidtumoren selten. Die Wahrscheinlichkeit, Metastasen (malignes Karzinoid) zu haben, korreliert mit der Größe des Tumors; diejenigen, die größer als 2 cm sind, haben eine Wahrscheinlichkeit von 60 % bis 80 %, Metastasen zu haben, und diejenigen, die kleiner als 1 cm sind, haben eine Wahrscheinlichkeit von weniger als 2 %, Metastasen zu haben. Daher können kleine rektale Karzinoidtumoren normalerweise erfolgreich durch einfache Exzision entfernt werden, aber die größeren Tumoren (größer als 2 cm) erfordern eine umfangreichere Operation, die die Entfernung eines Teils des Rektums beinhalten kann.
Es gibt drei Arten von Karzinoidtumoren im Magen; Typen I, II und III.
Magenkarzinoide vom Typ I die 75 % der Magenkarzinoide ausmachen, sind typischerweise kleiner als 1 cm und in der Regel gutartig. Es können mehrere Tumore im ganzen Körper des Magens verstreut sein. Sie entwickeln sich typischerweise bei Patienten mit perniziöser Anämie oder chronisch atrophischer Gastritis, Zuständen, bei denen der Magen keine Säure mehr produziert. Der Säuremangel führt dazu, dass die Zellen im Magen, die das Hormon Gastrin produzieren, große Mengen an Gastrin ins Blut absondern. (Gastrin ist ein Hormon, das normalerweise vom Körper produziert wird, um die Magensäure zu stimulieren. Die Säure im Magen stoppt die Produktion von Gastrin. Bei perniziöser Anämie oder chronisch atrophischer Gastritis führt der Säuremangel zu einer erhöhten Produktion von Gastrin.) Gastrin stimuliert zusätzlich zur stimulierenden Säure auch das Wachstum von enterochromaffinen Zellen im Magen zu gutartigen karzinoiden Tumoren. Die Behandlung von karzinoiden Tumoren vom Typ I umfasst Medikamente wie Medikamente vom Somatostatin-Typ, die die Produktion von Gastrin stoppen, oder die operative Entfernung des gastrinproduzierenden Teils des Magens.
Magenkarzinoide vom Typ II sind äußerst selten und wachsen sehr langsam mit einer geringen Wahrscheinlichkeit, bösartig zu werden. Sie treten bei Patienten mit einer seltenen genetischen Störung namens MEN (multiple endokrine Neoplasien) Typ I auf. Diese Patienten haben Tumore in anderen endokrinen Drüsen wie der Hypophyse, der Nebenschilddrüse und der Bauchspeicheldrüse.
Magenkarzinoide vom Typ III neigen dazu, größer als 3 cm zu sein und neigen dazu, sporadisch (ein oder zwei gleichzeitig auftretend) im ansonsten normalen Magen (ohne das Vorhandensein von perniziöser Anämie oder chronisch atrophischer Gastritis) aufzutreten. Typ-III-Tumoren sind normalerweise bösartig und neigen dazu, tief in die Magenwand einzudringen und zu metastasieren. Typ-III-Tumoren können lokale Symptome wie Bauchschmerzen und Blutungen sowie Symptome aufgrund eines Karzinoidsyndroms verursachen. Magenkarzinoide vom Typ III erfordern normalerweise eine chirurgische Entfernung des Magens sowie der umgebenden Lymphknoten.
Kolonkarzinoide treten typischerweise im rechten Dickdarm (Kolon aufsteigend und rechte Hälfte des Querkolons) auf. Wie die Karzinoide des Dünndarms werden auch die Karzinoide des Dickdarms oft spät entdeckt. So beträgt die durchschnittliche Größe der Tumoren zum Zeitpunkt der Diagnose 5 cm und Metastasen sind bei zwei Drittel der Patienten vorhanden. Das Karzinoidsyndrom ist bei Kolonkarzinoidtumoren selten.
Indem ich auf „Senden“ klicke, stimme ich den Geschäftsbedingungen und der Datenschutzrichtlinie von MedicineNet zu. Ich stimme auch zu, E-Mails von MedicineNet zu erhalten, und ich verstehe, dass ich mich jederzeit von MedicineNet-Abonnements abmelden kann.
Hausärzte, einschließlich Hausärzte und Internisten, werden oft zuerst das Vorliegen des Karzinoidsyndroms vermuten. Konsultationen sowohl mit Gastroenterologen als auch mit medizinischen Onkologen werden in Kürze folgen. Bei Verdacht auf einen Primärtumor im Magen-Darm-Trakt werden häufig Allgemeinchirurgen und chirurgische Onkologen hinzugezogen, auch wenn bereits Metastasen in der Leber vorhanden sind.
Es gibt mehrere Aspekte bei der Diagnose von Karzinoidtumoren:
In der klinischen Praxis wird die Diagnose eines karzinoiden Tumors am häufigsten zufällig gestellt, wenn Tests und Verfahren für andere Zwecke durchgeführt werden. Karzinoide Tumore im Enddarm und Dickdarm werden beispielsweise zufällig bei einer Darmspiegelung zur Darmkrebsvorsorge, bei Eisenmangelanämie oder chronischem Durchfall gefunden. Karzinoidtumoren des Blinddarms werden gefunden, wenn Appendektomien wegen Symptomen einer Appendizitis durchgeführt werden. Karzinoide des Magens werden zufällig entdeckt, wenn Endoskopien des oberen Gastrointestinaltrakts wegen Symptomen eines Geschwürs, Dyspepsie, Bauchschmerzen oder Anämie durchgeführt werden. Wenn diese zufällig gefundenen karzinoiden Tumoren kleiner als 1 cm sind, ist die Prognose gut, da die meisten von ihnen keine Metastasen gebildet haben und durch vollständige Exzision geheilt werden können.
Dünndarm-Bariumstudie. Es ist schwierig, die primären Karzinoide des Dünndarms frühzeitig zu finden, bevor sie bösartig werden und Metastasen bilden. Dünndarmtumoren (einschließlich karzinoider Tumore) sind schwierig zu diagnostizieren, da die traditionellen Barium-Röntgenaufnahmen des Dünndarms normalerweise nicht genau bei der Erkennung von Dünndarmtumoren sind, die den Darm noch nicht verstopfen. Außerdem kann der größte Teil des Dünndarms weder mit dem oberen Endoskop noch mit dem Koloskop erreicht werden. Daher werden Karzinoidtumoren des Dünndarms oft spät diagnostiziert, oft nachdem Lebermetastasen oder ein Karzinoidsyndrom aufgetreten sind.
Die Diagnose von Dünndarmtumoren wird einfacher, wenn Tumore entweder durch ihre Größe oder durch Narbenbildung um den Darm herum (fibrosierende Mesenteritis) einen Dünndarmverschluss verursachen, was zu einer Abknickung des Dünndarms führt (wie zuvor besprochen). Einfache Röntgenaufnahmen des Abdomens und Untersuchungen des Dünndarms mit Barium können beide eine Dünndarmobstruktion nachweisen, und ein computergestützter Axialtomographie-CT-Scan des Abdomens kann die ausgedehnte Narbenbildung einer fibrosierenden Mesenteritis nachweisen. Manchmal wird der karzinoide Tumor zum Zeitpunkt der Operation gefunden, die durchgeführt wird, um den Dünndarmverschluss zu lindern.
Kapselenteroskopie. In den letzten Jahren hat sich die Kapselenteroskopie weit verbreitet. Die Kapsel-Enteroskopie ist eine neuartige Technologie, bei der eine kleine Kapsel geschluckt wird, die eine Kamera und eine Lichtquelle enthält. Tausende von Bildern werden von der Kapsel aufgenommen, während sie durch den Dünndarm rollt, und diese Bilder werden an einen Empfänger übertragen, der um die Taille eines Patienten getragen wird. Viele Dünndarmerkrankungen (Geschwüre, Krebs, Lymphome, blutende Blutgefäße sowie karzinoide Tumore) wurden durch Kapsel-Enteroskopie entdeckt. Dieser Autor glaubt, dass mehr und mehr karzinoide Dünndarmtumoren frühzeitig entdeckt werden, wenn die Kapsel-Enteroskopie immer mehr Verbreitung findet.
Manchmal können die primären karzinoiden Tumore des Dünndarms oder Dickdarms durch nuklearmedizinisches Octreotid-Scanning oder durch CT-Scan des Abdomens diagnostiziert werden, aber häufiger sind diese Scans nützlicher beim Nachweis von Metastasen von karzinoiden Tumoren. (Siehe unten.)
Eine Möglichkeit, karzinoide Tumore zu diagnostizieren, besteht darin, zuerst das Karzinoid-Syndrom zu diagnostizieren und dann nach dem primären karzinoiden Tumor und seinen Metastasen zu suchen. Bei Patienten mit episodischen Anfällen von Flush, Durchfall und manchmal Keuchatmung kann die Diagnose des Karzinoidsyndroms bestätigt werden, indem die Ausscheidung von 5-Hydroxyindolessigsäure (5-HIAA) im über 24 Stunden gesammelten Urin gemessen wird.
Urin für 5-HIAA. Bei normalen, gesunden Personen wird ein Großteil der Aminosäure Tryptophan aus der Nahrung im Körper in Nikotinsäure umgewandelt. Karzinoidtumoren, die das Karzinoidsyndrom verursachen, wandeln den größten Teil des Tryptophans in Serotonin und 5-HIAA um. Normale Personen scheiden typischerweise weniger als 8 mg 5-HIAA in 24 Stunden aus. Patienten mit Karzinoidsyndrom können in 24 Stunden zwischen 100 und 2000 mg 5-HIAA ausscheiden. Wenn die Urinproben ordnungsgemäß gesammelt und die Tests ordnungsgemäß durchgeführt werden, liefert ein abnormal erhöhter 5-HIAA-Wert im Urin eine genaue Diagnose des Karzinoidsyndroms und sollte Bemühungen zur Suche nach den Karzinoidtumoren und ihren Metastasen veranlassen.
Bestimmte Lebensmittel und Medikamente können die Genauigkeit der 5-HIAA-Messungen im Urin beeinträchtigen, indem sie die 5-HIAA-Werte entweder fälschlicherweise erhöhen oder verringern. Diese Medikamente und Lebensmittel sollten 2 Tage vor und am Tag der Urinsammlung vermieden werden.
Lebensmittel, die 5-HIAA-Werte fälschlicherweise erhöhen, sind Avocados, Ananas, Bananen, Kiwis, Pflaumen, Auberginen, Walnüsse, Hickory-Nüsse und Pekannüsse. Zu den Medikamenten, die fälschlicherweise die 5-HIAA-Werte erhöhen, gehören Paracetamol (Tylenol), Robitussin, Phenobarbital, Ephedrin, Nikotin, Fluorouracil (Carac, Efudex, Fluoroplex) und Mesalamin (Asacol, Pentasa und Colazal).
Zu den Medikamenten, die die 5-HIAA-Werte fälschlicherweise senken können, gehören Aspirin, Heparin, Alkohol, Methyldopa, Imipramin, Isoniazid, Levodopa, Phenothiazine und MAO-Hemmer.
Chromogranin A. Chromogranin A ist ein Protein, das von karzinoiden Tumoren produziert wird. Es wird nicht so häufig wie 5-HIAA im Urin zur Diagnose des Karzinoidsyndroms verwendet, aber es wird von Ärzten zur Vorhersage der Prognose verwendet. Somit korreliert der Blutspiegel von Chromogranin A mit der Tumormenge im Körper (auch bekannt als Tumorlast). Patienten mit sehr hohen Chromogranin-A-Spiegeln haben eine schlechtere Überlebenszeit als Patienten mit niedrigeren Spiegeln.
Diagnose von karzinoiden Tumormetastasen
CT- und MRT-Scans. CT- und MRT-Scans (Magnetresonanztomographie) werden häufig zur Beurteilung von Bauchschmerzen, Gewichtsverlust, abnormalen Leberwerten und anderen Symptomen verwendet. Wenn bei diesen Scans Lebertumore oder abnormale Lymphknoten gefunden werden, kann eine Nadel in die Tumore oder Knoten eingeführt werden, um Gewebe für die Diagnose zu erhalten. Bei ausreichender Gewebeentnahme kann ein erfahrener Pathologe Karzinoidtumoren diagnostizieren, indem er das Gewebe unter dem Mikroskop untersucht. Leider stellen Tumore, die durch CT- und MRT-Scans gefunden werden, häufig Metastasen dar, wobei Lebermetastasen am häufigsten vorkommen. CT- und MRT-Scans sind nicht geeignet, um die primären Karzinoidtumoren im Dünndarm oder Dickdarm zu erkennen, wenn sie noch klein und resezierbar sind.
Indium-111-Octreotid-Scans. Karzinoide Tumorzellen haben wie alle anderen Zellen Membranen, die ihren Inhalt umgeben. Die Zellen von ungefähr 90 % der karzinoiden Tumoren haben Membranen, die mit Rezeptoren für ein Hormon namens Somatostatin bedeckt sind. Somatostatin bindet an diese Rezeptoren. Octreotid ist eine Chemikalie, die Somatostatin ähnelt und daher auch an die Rezeptoren bindet. Wenn radioaktives Indium 111-markiertes Octreotid in die Vene eines Patienten injiziert wird, bindet das radioaktive Octreotid an die Membran von karzinoiden Tumoren. Wenn der Patient unter eine Nuklearkamera gelegt wird, erscheinen die Karzinoidtumoren als helle Flecken auf dem Scan. Dieser Octreotid-Scan ist sehr genau (genauer als CT- und MRI-Scans) beim Nachweis von Leber- und anderen Metastasen von Karzinoidtumoren und auch genauer als CT-Scans und MRI-Scans zum Nachweis von primären Karzinoidtumoren. Patienten mit karzinoiden Tumoren, die auf Octreotid-Scans erscheinen, sprechen ebenfalls eher auf eine Behandlung mit Octreotid an. (Siehe unten.) Manchmal werden beim Octreotid-Scan zusätzliche karzinoide Tumoren in der Leber und den Lymphknoten gefunden, die auf dem CT-Scan nicht zu sehen sind.
Indium111-Octreotid-Scans haben Einschränkungen. Die Erkennungsrate von primären Karzinoiden durch den Octreotid-Scan beträgt immer noch nur 60 %. Scans können normalerweise keine primären Karzinoidtumoren erkennen, die kleiner als 1 cm sind. Scans können auch keine karzinoiden Tumore erkennen, die keine Somatostatinrezeptoren haben oder Rezeptoren haben, die Octreotid nicht binden. Es gibt zwei weitere nukleare Scans (PET-Scans und radioaktiver MIBG-Scan), die in Verbindung mit dem Octreotid-Scan verwendet werden können, was die Genauigkeit verbessern kann, aber die Erfahrung mit diesen beiden Scans ist begrenzt.
In der Praxis werden CT-, MRT- und Octreotid-Scans oft in Kombination verwendet, um Karzinoidtumoren zu erkennen, oft mit Genauigkeitsraten von fast 90 %. Die genaue Identifizierung aller Stellen des karzinoiden Tumors hat wichtige Implikationen für die Behandlung. Wenn zum Beispiel nur Lebermetastasen gefunden werden, kann der Patient möglicherweise durch eine chirurgische Resektion sowohl des Primärtumors als auch der Lebermetastasen behandelt werden. Wenn karzinoide Tumormetastasen sowohl in der Leber als auch in anderen Organen gefunden werden, ist der Patient kein guter Kandidat für eine Operation.
Knochenscan. Bei etwa 10 % der Patienten mit karzinoiden Tumoren metastasiert der Tumor in die Knochen und kann Knochenschmerzen verursachen. Knochenscans mit radioaktivem Phosphat sind genau, um diese Knochenmetastasen zu erkennen.
Karzinoidtumoren können gutartig (nicht krebsartig) oder bösartig (krebsartig) sein. Gutartige karzinoide Tumoren sind typischerweise klein (weniger als 1 cm). Sie können normalerweise vollständig entfernt werden und kommen in den meisten Fällen nicht wieder. Zellen von gutartigen karzinoiden Tumoren breiten sich nicht auf andere Teile des Körpers aus. Gutartige karzinoide Tumoren verursachen in der Regel keine Symptome und werden häufig zufällig während einer flexiblen Sigmoidoskopie oder einer Endoskopie des oberen Gastrointestinaltrakts gefunden.
Krebsartige oder bösartige karzinoide Tumoren sind zum Zeitpunkt der Diagnose typischerweise groß (größer als 2 cm). Zellen dieser bösartigen Tumore können Gewebe und Organe in der Nähe des Tumors befallen und schädigen. Darüber hinaus können sich bösartige Zellen lösen und in den Blutkreislauf oder das Lymphsystem gelangen und sich ausbreiten, um neue Tumore in anderen Teilen des Körpers zu bilden. (Die entfernten Tumoren werden Metastasen genannt.) Häufige Stellen für Karzinoidmetastasen sind Lymphknoten, Leber, Lunge, Knochen und Haut.
Bei karzinoiden Tumoren mit einer Größe zwischen 1,0 und 2,0 cm liegt die Wahrscheinlichkeit, dass sie zum Zeitpunkt der Diagnose krebsartig sind, bei etwa 10 %.
Karzinoide Tumoren wachsen typischerweise langsam. Sie wachsen viel langsamer als andere Krebsarten wie Dickdarm-, Bauchspeicheldrüsen-, Leber- und Lungenkrebs. Viele kleine karzinoide Tumoren verursachen keine Symptome und sind nicht tödlich; sie werden zufällig bei der Obduktion gefunden. Auch Patienten mit größeren bösartigen Karzinoidtumoren (mit oder ohne Metastasierung) können Jahre oder Jahrzehnte mit guter Lebensqualität überleben. Dies gilt insbesondere für moderne Behandlungen zur Kontrolle des Karzinoidsyndroms und zur Kontrolle des Wachstums von Karzinoidtumoren. Es gibt jedoch eine seltene Form von karzinoiden Tumoren, sogenannte Adenokarzinome, die aggressiver sind als die typischen bösartigen karzinoiden Tumore und eine schlechte Prognose haben. Ein erfahrener Pathologe kann Adenokarzinom-Tumoren identifizieren, indem er das Gewebe des Tumors unter einem Mikroskop untersucht.
Es gibt viele Optionen für die Behandlung von Karzinoiden:
Da karzinoide Tumoren in Größe, Malignitätspotential, Prognose, Ausmaß der Metastasierung und Symptome sehr unterschiedlich sind, sollte die Behandlung individuell angepasst werden. Da das Karzinoidsyndrom und metastasierende Karzinoidtumoren selten und ihre Behandlungen komplex sind, sollten die meisten Patienten von einem Team von Ärzten – Gastroenterologen, Onkologen, Radiologen, Kardiologen und Chirurgen – in medizinischen Zentren behandelt werden, die über Erfahrung und Ausrüstung zur Behandlung von Karzinoidtumoren verfügen .
Einige Patienten mit inoperablen Karzinoidtumoren haben möglicherweise weder lokale Symptome noch das Karzinoidsyndrom. Diese Patienten können ohne Operation oder Medikamente beobachtet werden, da karzinoide Tumore langsam wachsen und die Patienten möglicherweise über einen längeren Zeitraum keine Symptome entwickeln.
Eine Operation wird zur 1) kurativen Resektion, 2) Linderung von Symptomen wie Dünndarmverschluss oder Darmblutung und 3) Verringerung der Größe von Tumoren, die nicht vollständig resezierbar sind, ein Prozess, der als Tumordebulking bezeichnet wird, verwendet, um die Tumorlast zu verringern und zu verringern die von den Tumoren produzierte Hormonmenge.
Kleine rektale Karzinoidtumoren sind in der Regel gutartig und können zur Heilung oft vollständig entfernt werden. Magenkarzinoide vom Typ 1 sind in der Regel ebenfalls gutartig und können häufig zur Heilung entfernt werden. Kleine karzinoide Tumoren des Blinddarms werden normalerweise zum Zeitpunkt der Appendektomie entfernt und geheilt.
Carcinoid tumors of the small intestine and the colon often are large and have already metastasized at the time of diagnosis. Most patients with metastases are not candidates for a surgical cure because surgery cannot completely remove the entire tumor. Occasionally, a patient may have a solitary metastasis confined to a portion of the liver. Such patients can be treated with surgical resection of the primary tumor and resection of that part of the liver containing the tumor (partial hepatectomy). There are a limited number of patients with multiple metastases that are confined to the liver. Partial hepatectomy cannot be performed in these patients because of the multiple locations of the tumors. A small number of these patients have been treated successfully with liver transplantation.
Cryotherapy, radiofrequency ablation, and hepatic artery embolization all are techniques for debulking unresectable tumors (mainly liver metastasis) to decrease tumor burden and to treat the carcinoid syndrome. Effective debulking can improve the carcinoid syndrome and also prolong survival. Probes that freeze (cryotherapy) or deliver radiofrequency waves (RF ablation) can be inserted into the liver to debulk the liver of metastases from carcinoid tumors. Hepatic artery embolization involves blocking the arterial blood supply to carcinoid tumors (using oil-gelatin sponge particles) in the liver followed by chemotherapy to debulk the remaining liver tumors. Alternatively, radioactive microspheres can be injected into hepatic arteries to destroy liver tumors.
Interferon is a substance that inhibits the replication of some viruses and the growth of some tumors. Interferon has been used to treat patients with chronic hepatitis B and C. Interferon also has been found to arrest the growth of carcinoid tumors in some patients. Interferon has significant side effects, however.
Chemotherapy has been used alone or in combinations with other therapies to treat carcinoid tumors with metastases. The agents used include 5-fluorouracil (5-FU), cyclophosphamide, streptozotocin, and doxorubicin. The tumors do not frequently respond to treatment (a response is seen in under 20% of tumors), and the duration of response usually is only a few months. The side effects and toxicity of chemotherapy can be high.
External radiation has been used to alleviate pain due to the presence of metastases from carcinoid tumors in the spine. It also may reduce the size of the tumor in the spine. External radiation usually is not effective in treating tumors within the liver. Radioactive isotopes have also been used to palliatively treat metastatic carcinoid tumors with some benefit reported, although the series of patients so treated have been limited.
Medications for the control of the carcinoid syndrome
The most important treatment modality for carcinoid syndrome is octreotide, a synthetic hormone similar in structure to the naturally-occurring hormone, somatostatin. Somatostatin is widely distributed in the body where it can inhibit the secretion of many other hormones including growth hormone, insulin, and gastrin. It exerts its action by binding to specific receptors on the membranes of cells that produce and release hormones and chemical substances. Octreotide, like somatostatin, binds to receptors on the cells of carcinoid tumors and inhibits the manufacture and release of tumor hormones. Octreotide is very effective in controlling the symptoms of flushing and diarrhea that are part of the carcinoid syndrome. Octreotide has been found to reduce the excretion of 5-HIAA in some patients. Octreotide also has been found to slow the growth of carcinoid tumors, and, in a few patients, even reduce the size of the tumors and their metastases. Treatment with octreotide before surgery is important to prevent life-threatening carcinoid crises in patients with carcinoid syndrome undergoing surgery. Some doctors are advocating using octreotide even in patients without carcinoid syndrome to control the growth of the carcinoid tumors.
Octreotide generally is well tolerated. Side effects include nausea, headache, dizziness, abdominal pain, diarrhea, elevated blood sugar levels, and gallstones. The major drawback of octreotide is the need to inject it under the skin three times daily. Other longer-acting synthetic hormones resembling somatostatin (for example, lanreotide) can be given intramuscularly every 2 to 4 weeks.
Patients with carcinoid syndrome should take vitamin supplements, especially nicotinic acid since carcinoid tumors can cause a deficiency of nicotinic acid. In some patients, diarrhea caused by the carcinoid syndrome may respond to Imodium, Lomotil, ondansetron (Zofran), or cyproheptadine (Periactin). Patients also should avoid alcohol, spicy foods, physical stress, and ephedrine-containing medications such as nasal decongestants to avoid the precipitation of carcinoid syndrome by the release of hormones and chemical substances from the tumor. Patients with chronic diarrhea should take minerals supplements as well as vitamins since any cause of chronic diarrhea can lead to deficiencies of minerals.
A patient may be cured if a primary carcinoid tumor is discovered without carcinoid syndrome and is completely removed surgically. On rare occasions, a bronchial carcinoid may be found to be causing carcinoid syndrome without metastases being present. Removal of a bronchial carcinoid tumor can be curative, even if carcinoid syndrome is present.
A patient with the carcinoid syndrome and a primary carcinoid tumor which arose in the gastrointestinal tract has metastatic disease and is generally incurable. Nevertheless the clinical course of the disease is often considered slow or indolent.
 Typhus
Typhus
 Was sind die Symptome eines Schubs einer Divertikulitis?
Was sind die Symptome eines Schubs einer Divertikulitis?
 Lupus:Wie man den Autoimmunprozess ausschaltet und auf natürliche Weise heilt
Lupus:Wie man den Autoimmunprozess ausschaltet und auf natürliche Weise heilt
 Gute Nachrichten für RDS-Betroffene, da Forscher „Darmjucken“ feststellen
Gute Nachrichten für RDS-Betroffene, da Forscher „Darmjucken“ feststellen
 Barrett-Ösophagus
Barrett-Ösophagus
 5 Gründe, Kohlenhydrate für die Darmgesundheit NICHT zu reduzieren
5 Gründe, Kohlenhydrate für die Darmgesundheit NICHT zu reduzieren
 Wie ernst ist Morbus Crohn?
Was ist Morbus Crohn? Morbus Crohn an und für sich ist normalerweise nicht lebensbedrohlich, obwohl er schwere oder tödliche Komplikationen verursachen kann, die umfassen Darmverschluss, Fisteln, A
Wie ernst ist Morbus Crohn?
Was ist Morbus Crohn? Morbus Crohn an und für sich ist normalerweise nicht lebensbedrohlich, obwohl er schwere oder tödliche Komplikationen verursachen kann, die umfassen Darmverschluss, Fisteln, A
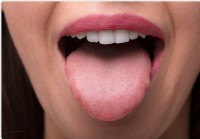 Mikroben auf der Zunge könnten zur Diagnose von Bauchspeicheldrüsenkrebs verwendet werden
Forscher der Medizinischen Fakultät der Zhenjiang-Universität, China hat herausgefunden, dass eine Störung der mikrobiellen Zusammensetzung des Zungenbelags als Biomarker für Bauchspeicheldrüsenkrebs
Mikroben auf der Zunge könnten zur Diagnose von Bauchspeicheldrüsenkrebs verwendet werden
Forscher der Medizinischen Fakultät der Zhenjiang-Universität, China hat herausgefunden, dass eine Störung der mikrobiellen Zusammensetzung des Zungenbelags als Biomarker für Bauchspeicheldrüsenkrebs
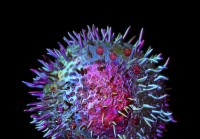 Die Rolle der Mastzellen für unsere Gesundheit
Mastzellen sind Zellen, die im Bindegewebe unseres Körpers als Teil unseres Immunsystems vorkommen. Mastzellen sind besonders prominent in Geweben unseres Körpers, die mit unserer Außenwelt interagier
Die Rolle der Mastzellen für unsere Gesundheit
Mastzellen sind Zellen, die im Bindegewebe unseres Körpers als Teil unseres Immunsystems vorkommen. Mastzellen sind besonders prominent in Geweben unseres Körpers, die mit unserer Außenwelt interagier