Alosetron (Lotronex) è stato approvato per la commercializzazione dalla FDA nel febbraio 2000, ma è stato ritirato dal mercato nel novembre 2000 a causa di gravi effetti collaterali gastrointestinali potenzialmente letali. Nel giugno 2002, è stato nuovamente approvato dalla FDA per la commercializzazione, ma in modo limitato, nell'ambito di un programma sponsorizzato da un'azienda farmaceutica per la gestione dei rischi associati al trattamento. L'uso di alosetron è consentito solo tra le donne con sindrome dell'intestino irritabile (IBS) grave, predominante nella diarrea, che non hanno risposto al trattamento convenzionale per l'IBS.
Di seguito è riportata la dichiarazione ufficiale originale della FDA rilasciata quando alosetron è stato originariamente ritirato.
-- Redattore medico, MedicineNet.com
28 novembre 2000--GLAXO WELLCOME DECIDE DI RITIRARE LOTRONEX DAL MERCATO Glaxo Wellcome, di Research Triangle Park, NC, ha informato la FDA che ritirerà volontariamente le compresse di Lotronex (alosetron cloridrato) dal mercato. Lotronex è un farmaco su prescrizione approvato per il trattamento della sindrome dell'intestino irritabile (IBS) nelle donne. La FDA consiglia ai pazienti che assumono Lotronex di contattare i propri operatori sanitari per discutere le alternative terapeutiche.
L'azione della società segue un incontro tenutosi oggi con la Food and Drug Administration (FDA) in cui l'agenzia ha discusso con Glaxo Wellcome le opzioni di gestione del rischio che includevano la limitazione della distribuzione del farmaco o il ritiro dal marketing.
L'azione di oggi segue le analisi della FDA sulle segnalazioni post-marketing di eventi avversi gravi, che includevano 5 segnalazioni di morte in pazienti che assumevano Lotronex.
In particolare, la FDA è preoccupata per i casi segnalati di danni intestinali risultanti dalla riduzione del flusso sanguigno all'intestino (colite ischemica) e dall'intestino gravemente ostruito o rotto (complicazioni di grave costipazione).
Al 10 novembre 2000, la FDA aveva ricevuto ed esaminato un totale di 70 casi di gravi eventi avversi post-marketing, inclusi 49 casi di colite ischemica e 21 casi di grave costipazione. Dei 70 casi, 34 hanno portato al ricovero in ospedale senza intervento chirurgico, 10 hanno portato a procedure chirurgiche e tre hanno provocato la morte. La FDA ha ricevuto due ulteriori segnalazioni di morte che l'agenzia non ha classificato come casi di colite ischemica o gravi complicanze della stitichezza.
La FDA ha monitorato da vicino il farmaco dall'approvazione del 9 febbraio 2000. Prima dell'approvazione, quattro casi di colite ischemica sono stati osservati negli studi clinici e sono stati discussi in una riunione del novembre 1999 del Comitato consultivo sui farmaci gastrointestinali della FDA. Questi casi erano transitori, di natura da lieve a moderata e reversibili con l'interruzione del farmaco.
Tra l'approvazione e il 1 giugno 2000, la FDA ha ricevuto sette segnalazioni post-marketing di gravi complicanze della stitichezza. Ciò ha comportato il ricovero di sei pazienti, tre dei quali hanno richiesto un intervento chirurgico. Nello stesso periodo, la FDA ha ricevuto otto segnalazioni post-marketing di colite ischemica. Ciò ha comportato quattro ricoveri, quattro procedure endoscopiche e nessun intervento chirurgico.
Il 27 giugno 2000, la FDA ha convocato una riunione del comitato consultivo pubblico in cui sono state discusse le opzioni di gestione del rischio in risposta alle segnalazioni di eventi avversi gravi. Nessun decesso è stato segnalato fino a quella data. Il consenso dei membri del comitato consultivo era che sia i medici che i pazienti dovevano essere informati degli eventi avversi potenzialmente gravi associati a Lotronex.
Dopo l'incontro, la FDA ha aggiornato l'etichettatura degli operatori sanitari per Lotronex e ha richiesto allo sponsor del farmaco, Glaxo Wellcome, di distribuire una guida ai farmaci che avvertisse direttamente i pazienti dei rischi associati al farmaco. Inoltre, su richiesta della FDA, Glaxo Wellcome ha emesso lettere "Gentile professionista sanitario" e "Gentile farmacista" per informare questi gruppi delle nuove importanti informazioni.
La FDA ha continuato a ricevere segnalazioni di eventi avversi gravi di colite ischemica e complicanze della stitichezza associate a Lotronex. Inoltre, la FDA ha ricevuto segnalazioni di morte e complicanze più gravi di colite ischemica che hanno richiesto trasfusioni di sangue o interventi chirurgici.
Dopo aver completato le sue recenti analisi dei 70 casi, il punto di vista della FDA sulle opzioni includeva il ritiro dall'immissione in commercio o un programma limitato di distribuzione dei farmaci. Il programma limitato di distribuzione dei farmaci fornirebbe:(1) l'uso sicuro di Lotronex in pazienti adeguatamente informati, (2) l'accesso continuo a Lotronex da parte di pazienti con IBS gravemente debilitati in condizioni strettamente monitorate e (3) la ricerca clinica continua sui benefici, i rischi, e un uso sicuro e appropriato di Lotronex. La FDA ha riconosciuto che gli altri trattamenti disponibili per l'IBS possono offrire un sollievo inadeguato da una condizione che può essere gravemente invalidante per alcuni pazienti.
Al termine della riunione odierna, Glaxo Wellcome ha informato la FDA che ritirerà volontariamente Lotronex dal mercato.
Per ulteriori informazioni su questo argomento, visitare la pagina Web Lotronex Information creata dal Center for Drug Evaluation and Research della FDA. L'URL è www.fda.gov/cder/drug/infopage/lotronex/lotronex.htm.
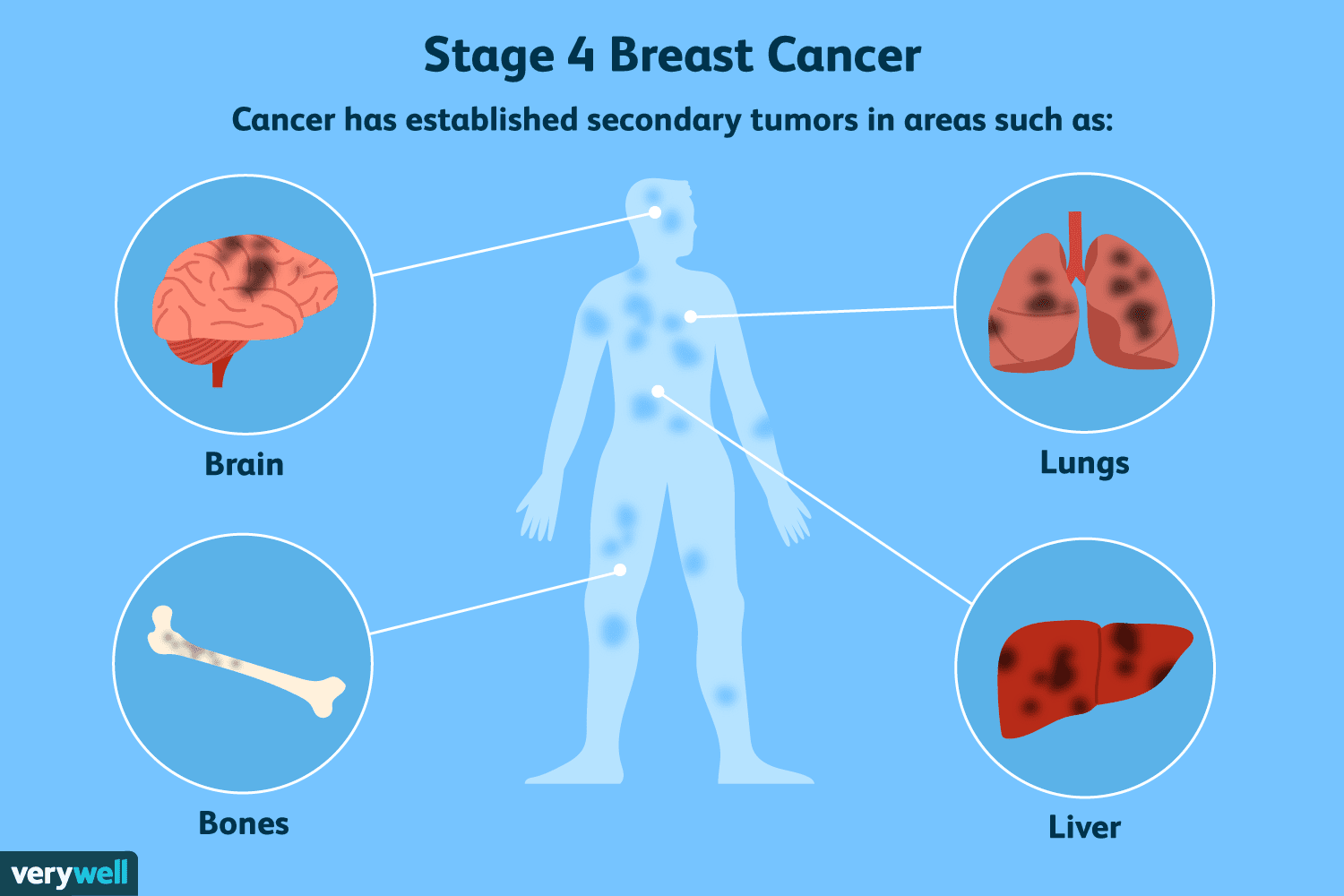 Una panoramica del cancro al seno in stadio 4
La fase 4 è la forma più avanzata di cancro al seno. Viene anche definito carcinoma mammario metastatico perché il tumore si sarà diffuso (metastatizzato) dal seno ad altre parti del corpo, come ossa,
Una panoramica del cancro al seno in stadio 4
La fase 4 è la forma più avanzata di cancro al seno. Viene anche definito carcinoma mammario metastatico perché il tumore si sarà diffuso (metastatizzato) dal seno ad altre parti del corpo, come ossa,
 Costipazione
La stitichezza cronica, a lungo termine o ricorrente è più difficile da trattare rispetto alla comune stitichezza semplice. Le persone che sono stitiche possono sperimentare uno o più di... ridotta
Costipazione
La stitichezza cronica, a lungo termine o ricorrente è più difficile da trattare rispetto alla comune stitichezza semplice. Le persone che sono stitiche possono sperimentare uno o più di... ridotta
 Sconfiggi Summer Bloat
Lora legale significa affrontare il gonfiore estivo? È comune. Dato che abbiamo così poco tempo per goderci davvero il bel tempo in Canada, appendiamo i grembiuli, trascorriamo il tempo nei patii, fac
Sconfiggi Summer Bloat
Lora legale significa affrontare il gonfiore estivo? È comune. Dato che abbiamo così poco tempo per goderci davvero il bel tempo in Canada, appendiamo i grembiuli, trascorriamo il tempo nei patii, fac