P27Kip1, regulert av glykogen syntase kinase-3β, resulterer i HMBA-indusert differensiering av humane magecancerceller
P27
Kip1, regulert av glykogen syntase kinase-3β, resulterer i HMBA-indusert differensiering av menneskelige mage kreftceller
Abstract
Bakgrunn
Magekreft er den nest vanligste årsaken til global kreftrelatert dødelighet . Selv dedifferentiation spår dårlig prognose i magekreft, den molekylære mekanismen underliggende dedifferentiation, noe som kan gi grunnleggende innsikt i svulst utvikling og progresjon, har ennå ikke klarlagt. Videre den molekylære mekanismen bak effekten av heksa bisacetamide (HMBA), en nylig oppdaget differensiering indus krever etterforskning og det er ingen rapporterte studier om effekten av HMBA på magekreft.
Metoder
Basert på resultatene av FACS-analyse, ble nivåene av proteiner involvert i cellesyklusen eller apoptose bestemt ved anvendelse av western blotting etter en enkelt behandling og sekvensielle kombinasjoner av HMBA og LiCl. GSK-3β og protonpumpe ble undersøkt ved western blotting etter opp-regulerer Akt uttrykk ved Ad-Akt infeksjon. For å undersøke effekten av HMBA på protein lokalisering og aktiviteter av GSK-3β, CDK2 og CDK4, kinasebestemmelsene immunoprecipitation og western blotting ble utført. I tillegg ble det nordlige blotting og RNase beskyttelse analyser utført for å bestemme den funksjonelle konsentrasjonen av HMBA.
Resultater
HMBA økt p27Kip1 uttrykk og indusert cellesyklus arrest i forbindelse med mage epitelcelledifferensiering. I tillegg er behandling av gastriske-avledede celler med HMBA-indusert G0 /G1 rest og opp-regulering av protonpumpe, en markør for magekreft differensiering. Videre behandling med HMBA øket ekspresjon og aktivitet av GSK-3β i kjernen, men ikke cytosol. HMBA nedsatt CDK2-aktivitet og indusert p27Kip1 uttrykk, som kan bli reddet ved hemming av GSK-3β. Videre økte HMBA p27Kip1 binding til CDK2, og dette ble opphevet av GSK-3β hemming.
Konklusjoner
Resultatene som presenteres her, tyder på at GSK-3P funksjoner ved å regulere p27Kip1 montering med CDK2, og dermed spille en avgjørende rolle i G0 /G1 arrest i forbindelse med HMBA gastrisk epitelcelledifferensiering.
nøkkelord
HMBA magekreft GSK-3β Bakgrunn
mage~~POS=TRUNC kreft~~POS=HEADCOMP er en av de vanligste kreftformene i verden, og ofte utvikler resistens mot kjemoterapi og strålebehandling behandlinger. Derfor har kombinasjonsterapi blitt foreslått å takle sykdommen bedre og for å redusere sannsynligheten for å utvikle resistens [1]. Heksametylen bisacetamide (HMBA), en hybrid polar forbindelse (HPC) opprinnelig utviklet som en differensieringsinduserende middel [2-6], bevirker gastrisk celle gjendifferensiering [7-9].
I magesekken, stamceller i proliferativ cellesone av eidet regionen i de gastriske kjertler differensiere og gi opphav til forskjellige celletyper [10, 11]. Når den første tumorigen hendelsen finner sted, avhenger videre tumorprogresjon på arten av den innledende hendelsen og utviklingsstadiet av cellen som vedvarende det og ytterligere mutasjoner som kan forekomme. Konstant spredning er en viktig funksjon av stamceller, og i gastrointestinale vev mutasjoner er sannsynligvis resultere i utvidelse av endrede stamceller, noe som øker sannsynligheten for ytterligere mutasjoner og tumorprogresjon [12]. Derfor er rettet mot gastrisk kreft stamceller er sannsynlig å være den mest effektive måten for behandling av magekreft. Omtrent 50% av den vestlige befolkningen utvikler metaplasi, et viktig steg i utviklingen av kreft [13], og trekker oppmerksomhet til trasé som styrer spredning og dermed celledifferensiering. Blant disse er TGF-b, MYB, Wnt og Hedgehog banene er av særlig betydning, med fremtredende i celle-skjebne spesifikasjon og mønster formasjon under embryogenese og voksen vev fornyelse. Klarlegging av komplekse tumor suppressor og gasssignalveier, som effekt differensiering modulering av overgangs /stamceller, vil være avgjørende for optimalisering av legemiddel å behandle magekreft.
For umodne mage cellene differensieres, de krever å bo i G1 fasen av cellesyklusen for en viss tidsperiode. Pattedyrcellesyklusen reguleres ved sekvensiell aktivering og inaktivering av en høyt konservert familie av cyklin-avhengige kinaser (CDK); progresjon gjennom tidlig til midten av G1 er avhengig av CDK4 og CDK6 muligens, mens progresjon gjennom sent, G1 og S-fasen krever aktivering av CDK2. Virksomheten til CDK kan hemmes ved binding av CDK hemmende proteiner inkludert Cip /Kip familie (p21 Waf1, p27 Kip1 og p57 Kip2) og INK4 familie (p15Ink4b, p16INK4a, p18Ink4c og p19Ink4d ). P27 Kip1 reguleres etter transcriptionally ved proteolytisk degradering. CDK2 binder seg til p27 Kip1 og fosforylerer det på treonin 187 [14], og HMBA-indusert gastrisk celledifferensiering er forbundet med den opp-regulering av p27 Kip1 [15, 16] og G0 /G1 arrest. Men det er få detaljerte studier om den molekylære mekanismen for HMBA og det har ikke vært rapportert om studier som undersøker effekten av HMBA på magekreft.
Som nedstrøms målet for phosphatidylinositol-3 kinase /Akt (PI3-kinase /Akt ) vei, regulerer GSK-3β celleproliferasjon og differensiering [17-20]. Samle bevis indikerer at hemmet GSK3p signalering, som fungerer i G1 å motta innspill fra flere signalering og utviklingsveier, oppstår i forbindelse med ulike kreft hos mennesker [21, 22]. GSK-3β har vært innblandet i flere biologiske prosesser fordi det fosforylerer et bredt spekter av underlag inkludert flere differensiering sjekkpunkter inkludert c-myc, snegl og PI3K [23]. Tidligere inhibering av PI3-kinase pathway ble vist å forbedre HMBA-mediert gastrisk celledifferensiering [8]. I denne studien ble rollen som GSK-3β i løpet av gastrisk celledifferensiering undersøkt ved å bruke den humane magecancercellelinje SGC7901, som viser en multipotent fenotype og representerer en velkarakterisert modell av mave differensiering. Resultatene antyder en bidragende rolle for GSK-3β i p27 Kip1 bane i løpet av gastrisk celledifferensiering indusert av HMBA.
Methods
Cellekultur
magekreft cellelinjen SGC7901 ble oppnådd fra cellebanken i den kinesiske Academy of Sciences og kultivert som tidligere beskrevet [24]. SGC7901-celler ble infisert med adenovirus som koder for aktivert form av Akt (Ad-Akt) eller den adenovirale vektor som koder for kontroll β-galaktosidase (β-gal) ved en multiplisitet av infeksjon (MOI) på 10 PFU /celle. Etter infeksjon med vektorer i en time, etterfulgt av erstatning av medium og inkubering i ytterligere 24 timer ble cellene behandlet i nærvær eller fravær av HMBA og protein og RNA ble ekstrahert for vestlig og Northern blotting, respektivt.
Materialer
HMBA, TSA, SB-415 286 og LiCl ble kjøpt fra Sigma Chemical Company, USA. Adenovirus vektorer som koder β-gal og Myristoylated aktive formen av Akt (Ad-Akt) ble kjøpt fra Cell BioLabs, USA. Vektoren koder for det katalytisk aktive mutant av GSK3P (HA-GSK-3βCA) ble kjøpt fra Addgene. Non-målretting kontroll siRNA og SMARTpool for målretting GSK-3β ble kjøpt fra Dharmacon, USA. Alle prober ble merket med en Biotin Random Prime DNA Merking Kit (Pierce).
Antistoffer
Rabbit anti-Akt, anti-fosfor-Akt (Ser473) og antiphospho-GSK-3β (Ser9) ble kjøpt fra Cell Signaling , USA. Mouse anti-GSK-3β, mus anti-p27 Kip1, mus anti-Top IIb og mus anti-p21 Waf1 ble kjøpt fra BD Biosciences, USA. Mouse anti-fosfor-GSK-3 (Tyr278 /Tyr216) ble hentet fra Upstate, USA. Kanin anti-spaltede PARP antistoff ble kjøpt fra Abcam, USA. Anti-proton pumpen ble kjøpt fra MBL International (USA). Polyklonale anti-CDK2, anti-CDK4, anti-α-tubulin og anti-caspase-3 ble oppnådd fra Santa Cruz Biotechnology (USA)
. Sub-cellulært protein utvinning og western blotting
Kjerne- og cytosoliske fraksjoner ble hentet ved hjelp av en NE-pER Atom og cytoplasmatiske Utvinning Reagenser kit (Pierce, USA). Cytosolisk protein (80 mg) eller kjerneprotein (20 mg) ble løst i en 10% polyakrylamid-gel og overført til PVDF-membraner som tidligere beskrevet [25]. Filtrene ble inkubert i en time ved romtemperatur i blottings oppløsning. Membraner ble inkubert over natten ved 4 ° C med primære antistoffer, etterfulgt av blotting med et pepperrot peroksidase-konjugert sekundært antistoff i en time, og visualisert ved hjelp av et ECL-påvisningssystem.
Northern blotting og RNase beskyttelses assays (RPA)
Total RNA ble fremstilt ved anvendelse av TRIzol reagens (Invitrogen). Prøvene ble kjørt på 1,2% agarose /formaldehyd-geler og overført til nitrocellulose som støttes. Membraner ble hybridisert med et biotin merket mage protonpumpe cDNA probe. Etter hybridisering med GAPDH-probe, en lastekontroll, ble membranene vasket og signaler ble detektert ved anvendelse av et ECL-påvisningssystem. RNase beskyttelse eksperimenter ble utført ved anvendelse av RPA-III-kit fra Ambion og RiboQuant flers RNase beskyttelse analysesystem ble anvendt for å påvise flere spesifikke mRNA. 32P-merkede antisense RNA prober ble utarbeidet etter de menneskelige apoptose HCC-2 og hCYC-1 Mal Stiller og hybridiseringer ble utført i henhold til produsentens protokoll.
Cell syklus analyse
Gastric kreftceller ble høstet ved hjelp av trypsin . Celler ble samlet opp, vasket to ganger med iskald PBS og fiksert i iskald 70% etanol. Etter å ha blitt vasket to ganger med iskald PBS, resuspendert i PBS inneholdende 100 U /ml RNase A og inkubert ved 37 ° C i 30 minutter, ble cellene farget med PI (20 mg /ml) og analysert ved bruk av FACScan (Becton Dickinson, San Jose, CA, USA) som tidligere beskrevet [25].
In vitro kinasebestemmelser
aktivitetene til CDK2, CDK4 og GSK-3β ble målt som tidligere beskrevet [26, 27]. I korte trekk ble CDK2, CDK4 eller GSK-3β immunopresipitert fra cytosoliske (100 mg protein) eller nukleær (25 mg protein) ekstrakter. Kinase-aktivitet ble målt ved å inkubere immunopresipitert CDK2, CDK4 eller GSK-3β i 40 ml kinasebuffer med 4 mg rekombinant snegle-protein (for å måle GSK-3β-assosiert kinaseaktivitet), 5 mg av histon H1 (for å måle CDK2-assosiert kinase aktivitet) eller retinoblastom protein (for å måle CDK4-assosiert kinase-aktivitet) ved 30 ° C i 30 minutter. Prøvene ble behandlet som beskrevet i tidligere rapporter [28].
Resultater
Hemming av GSK-3β demper HMBA-indusert cellesyklus arrest og SGC7901 celledifferensiering
SGC7901 celler akkumulert på G0 /G1 cellesyklus sjekkpunkt og differensiert i et enterocyte-lignende fenotype etter behandling med HMBA [29]. GSK-3β bidrar til inhibering av cellesyklusprogresjon i differensierende celler [20, 30]. Derfor, om GSK-3β spiller en rolle i HMBA-indusert SGC7901 cellesyklus inhibering ble undersøkt. Som vist i figur 1a, til behandling med HMBA induserte celler samle seg på G0 /G1 cellesykluskontrollpunktet. Behandling med litiumklorid (LiCl), som hemmer GSK-3β i et Mg 2 + konkurrerende måte, [31], økes andelen av celler i S-fasen. Behandling med en kombinasjon av LiCl og HMBA reversert HMBA-mediert G1 cellerest. Lignende resultater ble oppnådd etter behandling med SB-415286, er en potent inhibitor av GSK-3β [32] (Tilleggs fil 1). Disse resultater antyder at GSK-3β kan spille en rolle i HMBA-indusert G1 arrest. For å avgjøre om HMBA resulterte i celledød i løpet av 24 timer behandlingsperioden, ble protein ekstrahert å vurdere om det var økt PARP spalting og /eller aktiv caspase-3. Som vist i figur 1b, var det ingen økning i PARP spalting og aktiv caspase-3 til 48 timer etter HMBA behandling. En viktig tidlig begivenhet i den terminale differensiering av celler er deres tilbaketrekning fra cellesyklusen [6]. Siden GSK-3β er dokumentert til å spille en rolle i cellesyklus-stans [33], ble det postulert at hemming av GSK-3β kunne hemme differensiering. Derfor er virkningen av GSK-3P-inhibitorer for induksjon av HMBA-mediert gastrisk protonpumpeinhibitor uttrykk, en markør for gastrisk differensiering, ble undersøkt. SGC7901 celler ble forbehandlet med LiCl (figur 1c og 1d) eller SB-415286 (figur 1e og 1f) ved forskjellige konsentrasjoner i en time, og deretter behandlet med HMBA i 24 timer. LiCl hemmet HMBA-indusert gastrisk protonpumpeinhibitor ekspresjon i en doseavhengig måte. I samsvar med disse resultatene, SB-415 286 blokkert gastrisk protonpumpe protein og mRNA uttrykk, som ble indusert av HMBA. Tatt sammen indikerer disse resultater at GSK-3β spiller en viktig rolle i HMBA-mediert gastrisk celledifferensiering. Figur 1 Hemming av GSK-3β demper HMBA-indusert cellesyklus arrest og SGC7901 celledifferensiering. A, SGC7901-celler ble forbehandlet med eller uten 10 mM LiCl i 30 minutter, fulgt av kombinasjonsbehandling med 10 mM HMBA i 24 timer, fulgt med kvantifisering av DNA-innhold ved strømningscytometri. B, SGC7901-celler ble behandlet med HMBA (10 mM) i 24 timer eller 48 timer og ble fremstilt for western-blotting-analyse. C & E, SGC7901-celler ble forbehandlet med eller uten 10 mM LiCl eller 10 μMSB-415286 i en time, etterfulgt av kombinasjonsbehandling med 10 mMHMBA i 24 timer. protonpumpe-ekspresjon status ble bestemt ved hjelp av western-blotting-analyse. D &F, total-RNA ble ekstrahert fra cellene og Q-RT-PCR-analyse for protonpumpe-mRNA-ekspresjon ble utført. (Data representerer gjennomsnitt ± SD, * = p < 0,05 vs. kontroll; * = p < 0,05 vs. HMBA alene.)
Akt regulerer mage differensiering indusert av HMBA
GSK-3β inaktiveres når det. fosforyleres nedstrøms Akt [34]. Derfor ville det forutsies at aktivering av Akt med PI3-kinase vil være forbundet med hemming av GSK-3β og, senere, inhibering av gastrisk celledifferensiering. For å teste denne hypotesen, ble SGC7901 celler infisert med Ad-Akt eller en kontroll vektor. Infeksjon med Ad-Akt økt uttrykk av fosforylert Akt, Akt og fosforylert GSK-3β protein (figur 2a), i samsvar med tidligere resultater som viser at GSK-3P fungerer som et substrat av Akt. Som vist i Figur 2b, infeksjon av SGC7901 celler med Ad-Akt adenoviral vektor alene hadde ingen effekt på gastrisk protonpumpeinhibitor og mRNA-ekspresjon. Imidlertid infeksjon med Ad-Akt vektor resulterte i inhibering av gastrisk protonpumpeinhibitor mRNA-ekspresjon indusert av HMBA sammenlignet med HMBA og infeksjon av kontrollen (β-gal) adenovirus, noe som tyder på at signalisering gjennom de PI3-kinase /Akt sti regulerer gastrisk celle differensiering indusert av HMBA behandling. Figur 2 Akt regulerer mage differensiering indusert av HMBA. A, ble cytosol og kjerneproteiner ekstrahert fra celler behandlet som angitt, og løst på SDS-PAGE og blottet med anti-fosfo-Akt, -Akt, fosfo-GSK-3β, og GSK-3β, ved anvendelse av anti-α-tubulin og Topo IIβ som kontroll av cytosolisk og atomfraksjoner henholdsvis. B, protonpumpe-ekspresjon ble målt ved hjelp av western-blotting fulgt med overflod kvantifisering. (Data representerer gjennomsnitt ± SD, * = p < 0,05 vs. kontroll, † = p < 0,05 vs. HMBA alene.). C, Total RNA (40 ug) ble fraksjonert, overført til nitrocellulosemembraner og probet med et merket proton pumpe cDNA; blotter ble strippet og reprobed med GAPDH.
Behandling med HMBA økt uttrykk og aktivitet av GSK-3β i kjernen
å teste om GSK-3β var påvirket av HMBA behandling, GSK-3β aktivitet ble bestemt ved å måle fosforylering av rekombinant snegle, en godt karakterisert substrat av GSK-3β [35, 36]. GSK-3β ligger i den cytosoliske og nukleære avdelinger av celler, men hovedsakelig i cytoplasma i G1 fase. Derfor, nukleære og cytoplasmiske proteiner ble fraksjonert fra kontroll og HMBA-behandlede celler og kontrollert for GSK-3β aktivitet. HMBA behandling resulterte i en økning i aktiviteten av atom GSK-3β (figur 3a), og GSK-3β inhibering svekkede HMBA-mediert G1 rest, noe som indikerer en rolle for GSK-3β i HMBA-indusert cellesyklus-stans. Ser9 fosforylering av GSK-3β reduserer GSK-3β aktivitet, mens Tyr216 fosforylering øker GSK-3β aktivitet [37]. For å analysere mekanismene bak økt GSK-3β aktivitet forårsaket av HMBA behandling, Ser9-fosforylert og Tyr216-fosforylert GSK-3β protein ble bestemt ved hjelp av western blotting. HMBA behandling økte atom uttrykk nivåer av total GSK-3β og Tyr216-fosforylert GSK-3β uten at det påvirker deres uttrykk i cytosol (figur 3b). Interessant, HMBA behandlingen økte Ser9-fosforylert GSK-3β protein ekspresjon i både den cytosoliske og atomfraksjoner. Lignende resultater ble oppnådd etter behandling med andre HPCS, Saha og EMBA (tilleggsfiler 2). I tillegg HPC øket aktiviteten av GSK-3β i kjernen som demonstrert ved in vitro kinasebestemmelser
(Tilleggs fil 3). Disse resultatene tyder på at HPC øker atom GSK-3β aktivitet uavhengig av fosforylering ved Ser9. Figur 3 Behandling med HMBA øket ekspresjon og aktivitet av GSK-3β i kjernen. A, SGC7901 celler ble behandlet med (+) eller uten (-) 10 mMHMBA i 24 timer og høstes ved slutten av behandlingen. Cytosol og atom fraksjoner ble fremstilt og GSK-3β aktivitet ble analysert ved hjelp av in vitro-kinaseanalyse ved bruk av snegle-protein som substrat. Fosforylert Snail protein signalene ble densitometrically kvantifisert og uttrykt som fold-endring med hensyn til ubehandlede kontrollgrupper. B, SGC7901-celler ble behandlet med 10 mM HMBA i forskjellige tidsrom. Cytosoliske og kjernekraft proteinfraksjoner ble hentet ut og western blotting ble utført ved hjelp av antistoffer mot GSK-3β, fosfor-GSK-3β (Ser9), fosfor-GSK-3α /β (Tyr278 /Tyr216), α-tubulin eller Topo IIβ.
hemming av GSK-3β styrer HMBA-indusert CDK2 hemming
Progresjon gjennom G1 er avhengig CDK2 og CDK4 [38, 39]. For å bestemme hvorvidt GSK-3β regulering av HMBA-mediert G1 cellesyklusstans via CDK2 eller CDK4 hemming, SGC7901-celler ble forbehandlet med LiCl (figur 4a) eller SB-415286 (figur 4b), og deretter utsatt for kombinasjonsbehandling med HMBA i 24 timer før den immunoutfellingsstudier analyser ble utført på cellelysater under anvendelse av anti-CDK2 og anti-CDK4 antistoffer, henholdsvis. I tillegg ble CDK2 eller CDK4 aktivitet bestemt ved anvendelse av en in vitro-kinaseanalyse
. Behandling med HMBA alene hemmet CDK2 aktivitet (topp, venstre panel), men økt CDK4 aktivitet (øverst, høyre panel); hemming av GSK-3β hjelp LiCl eller SB-415286 betydelig svekket HMBA hemming av CDK2 aktivitet (nederst). Behandling med HMBA øket ekspresjon og aktivitet av GSK-3β i kjernen. Samlet utgjør disse resultatene tyder på at GSK-3β bidrar til HMBA-mediert G1 cellesyklus arrest gjennom hemming av CDK2. Figur 4 Hemming av GSK-3β styrer HMBA-indusert CDK2 hemming. SGC7901 celler ble forbehandlet med (+) eller uten (-) 10 mM LiCl (A) eller 10 mM SB-415286 (B) i 30 minutter, fulgt av kombinasjonsbehandling med 10 mM HMBA i 24 timer. Proteinekstrakter ble immunopresipitert med anti-CDK2 og anti-CDK4 antistoffer. Resulterende immunkompleksene ble analysert for CDK2-aktivitet ved anvendelse av histon H1 som substrat (øvre panel) eller for CDK4 aktivitet ved anvendelse av Rb som substrat (nedre panel). Fosforylert histon H1 eller Rb protein signalene ble kvantifisert densitometrically og uttrykt som fold-endring med hensyn til ubehandlede kontrollgrupper.
GSK-3β regulerer kjerne p27Kip1protein uttrykk
å undersøke mekanismene bak HMBA mediert G1 arrest og CDK2 hemming videre, cellesyklusregulerende mRNA uttrykk ble analysert ved hjelp av RPA analyser. Behandling med HMBA økt P27 Kip1 mRNA uttrykk, men redusert p53, p57, P15 (figur 5A høyre), cyclin A og cyclin D1 mRNA uttrykk (figur 5A venstre). Men behandling med LiCl økt p21 Waf1 mRNA uttrykk men ikke påvirke uttrykket nivåer av andre gener. Lignende resultater ble oppnådd når cellene ble behandlet med SB-415286 (Tilleggs fil 4). Disse resultater antyder at GSK-3β regulering av HMBA-mediert cellesyklus-stans ikke innebærer den transkripsjonelle regulering av cellesyklusrelaterte gener. Figur 5 mRNA ekspresjon av gener med hensyn til cellesyklusen. A, RNase beskyttelse analyser ble utført ved anvendelse av RNA fra SGC7901 celler behandlet med enten 10 mM HMBA, 20 mM LiCl, eller en kombinasjon av HMBA og LiCl i 24 timer, hybridisert med multi-prober for cellesyklusavhengig kinaseinhibitorer (A; hCC- 2) eller cykliner (B; hCYC-1). B, SGC7901-celler ble forbehandlet med (+) eller uten (-) 10 mM LiCl (til høyre) eller 10 mM SB-415286 (venstre) i 30 minutter, fulgt av kombinasjonsbehandling med 10 mM HMBA i 24 timer. Hele celle proteinekstrakter ble løst på SDS-PAGE, overført til PVDF membraner, og immunoblottet med antistoffer mot p27Kip1, p21Waf1, CDK2, eller β-aktin.
For å analysere mekanismene bak GSK-3β-forbundet cellesyklus arrest videre, ekspresjon av p21 Waf1 og p27 Kip1 proteiner i SGC7901 celler behandlet med HMBA i nærvær eller fravær av LiCl eller SB-415286 ble undersøkt. Tilsetting av LiCl (figur 5B høyre) eller SB-415 286 (figur 5B venstre) svekket induksjon av p27 Kip1 men ikke p21 Waf1 protein uttrykk, noe som tyder på at p27 Kip1 deltar i cellesyklus overganger regulert av GSK-3β. Når p27 Kip1 akkumuleres i kjernen det binder seg til CDK2, begrenser sin aktivitet, og til slutt induserer cellesyklus arrest. Videre HMBA (10 mM) øket p27 Kip1 protein ekspresjon i cytosol og kjernen mellom 0 til 24 timer etter behandlingen (figur 6a). HMBA (0-5 mm) økte p27 Kip1 uttrykk etter 24 timer i cytosoliske fraksjoner og etter 48 h HMBA (5-10 mm) økte p27 Kip1 uttrykk i kjernefysiske fraksjoner (Figur 6b). Tilsetting av LiCl (figur 6c) eller SB-415 286 (figur 6d) blokkert HMBA-økt p27 Kip1 atom uttrykk uten at det påvirker p27 Kip1 uttrykk i cytosol, noe som tyder spesifikk regulering av kjernefysisk p27 Kip1 uttrykk ved GSK-3β. For å demonstrere den rollen som GSK-3β i regulering av atom p27 Kip1 ekspresjon ytterligere, ble celler transfektert med siRNA rettet mot GSK-3β (figur 6e). RNAi-mediert hemming av GSK-3β ble bekreftet av immunoblotting og svekket atom p27 Kip1 induksjon HMBA uten å påvirke cytosolic p27 Kip1 induksjon. For å bekrefte den rollen som GSK-3β i regulering av atom p27 Kip1 ekspresjon, SGC7901-celler ble transfektert med en vektor som koder for aktivert form av GSK-3β (GSK-3β-CA) eller en tom kontrollvektor. Cytosol og nukleære proteiner ble hentet ut og western blotting ble utført for å bestemme p27 Kip1 uttrykk. Transfeksjon av SGC7901 celler med GSK-3βCA plasmid resulterte i økt p27 Kip1 i atomfraksjonen (figur 6F) uten å påvirke p27 Kip1 nivåer i cytosol. Over-ekspresjon av den aktive formen av GSK-3β ble bekreftet ved hjelp av Western-blotting og in vitro
kinase-analyser ved anvendelse av snegle-protein som substrat (figur 6G). Samlet utgjør disse resultatene indikerer at GSK-3β deltar i regulering av cellesyklus gjennom den konkrete reguleringen av atom p27 Kip1 protein uttrykk. Figur 6 Nuclear p27 Kip1 uttrykk modulert av GSK-3β. A & B, SGC7901-celler ble behandlet med HMBA (10 mM) i løpet av et tidsforløp (A) eller med forskjellige konsentrasjoner i 24 timer. Cytosoliske og kjernekraft proteinfraksjoner ble hentet ut og western blotting ble utført med antistoffer mot p27Kip1, α-tubulin eller Topo IIβ. Leir; D, SGC7901-celler ble forbehandlet med (+) eller uten (-) 20 mM LiCl (C) eller 10 mM SB-415286 (D) i 30 min, fulgt av kombinasjonsbehandling med 10 mM HMBA i 24 timer. Cytosol og nukleære proteiner ble ekstrahert for analyse av p27Kip1 proteinekspresjon. E, SGC7901 celler ble transfektert med siRNA rettet mot GSK-3β eller kontrollere siRNA. Tjuefire timer etter transfeksjon ble cellene behandlet med HMBA i ytterligere 24 timer. Cytosol og nukleære proteiner ble ekstrahert for analyse av p27Kip1 proteinekspresjon. Knockdown av GSK-3β ekspresjon ble bekreftet ved Western-blotting ved anvendelse av anti-GSK-3β antistoff. F, SGC7901 celler ble infisert med Ad-HA-GSK-3βS9A eller Ad-β-gal på en MOI på 10 PFU /celle. Etter 48 timers inkubering ble cytosol og kjerneprotein ekstrahert og western blotting utført ved anvendelse av anti-p27Kip1, anti-HA, og anti-GSK-3 antistoffer, henholdsvis ved anvendelse av anti-α-tubulin eller Topo IIβ som lasting kontroll. GSK-3P aktiviteter ble analysert ved hjelp av in vitro-kinaseanalyse ved bruk av snegle-protein som substrat (nedre panel). p27Kip1 signalene ble kvantifisert densitometrically og uttrykt som fold-endring med hensyn til a-tubulin eller TopIIβ.
GSK-3β regulerer p27Kip1binding til CDK2
HMBA økt p27 Kip1 protein uttrykk, hemmet CDK2 aktivitet og økt CDK4 aktivitet (figur 4). Utdrag fra kontroll eller HMBA-behandlede celler ble immunoutfelt å undersøke p27 Kip1 binding til CDK2 og CDK4. Som vist i figur 7a, HMBA behandling økt nivå av p27 Kip1 i kompleksene immunoutfelt med anti-CDK2, men ikke i komplekser immunoutfelt med anti-CDK4. Re-sentret filtrene med anti-CDK2 og anti-CDK4 antistoffer bekreftet at immunopresipitatene fra kontroll og HMBA-behandlede celler inneholdt de samme nivåer av CDK2 og CDK4. Derfor synes HMBA til å forårsake en selektiv økning i p27 Kip1 binding til CDK2. For å analysere om hemming av GSK-3β påvirker sammenslutning av p27 Kip1 med CDK2, SGC7901 celler ble forbehandlet med LiCl eller SB-415 286 og utsatt for kombinasjonsbehandling med HMBA i 24 timer; hel-celle-ekstrakter ble immunoutfelt. Behandling med GSK-3P-hemmere, LiCl (7b) eller SB-415 286 (figur 7c) blokkert p27 Kip1 binding til CDK2. Disse resultater antyder at GSK-3β er essensiell for HMBA-indusert økning i p27 Kip1 binding til CDK2. For å bekrefte den rollen som GSK-3β i regulering av p27 Kip1 tilknytning til CDK2, ble SGC7901-celler transfektert med en vektor som koder for aktivert form av GSK-3β eller Ad-β-gal. Hel-celle-protein ble ekstrahert og immunopresipitert. Som presentert i figur 7d, transfeksjon av SGC7901 celler med GSK-3β-CA vektor resulterte i et øket nivå av p27 Kip1 i kompleksene immunoutfelt med anti-CDK2, sammenlignet med transfeksjon av plasmid kontroll. Dette tyder på at GSK-3β er ikke bare nødvendig for HMBA-mediert p27 Kip1 binding til CDK2, men også tilstrekkelig til å øke sammenslutning av p27 Kip1 med CDK2 i SGC7901 celler. Figur 7 GSK-3β regulering av p27 Kip en tilknytning CDK2. A, SGC7901 celler ble behandlet med (+) eller uten (-) 10 mM HMBA i 24 timer. Proteinekstrakter ble immunopresipitert med anti-CDK2 og anti-CDK4 antistoffer. Normal kanin-IgG ble anvendt som kontroll. Den CDK2-eller CDK4-assosiert p27Kip1 i den resulterende immunkomplekser ble analysert ved western blotting ved hjelp av anti-p27Kip1 antistoff med anti-CDK2 eller -CDK4 antistoff som lasting kontroller. B &C, SGC7901-celler ble forbehandlet med (+) eller uten (-) 10 mM LiCl (B) eller 10 mM SB-415286 (C) i 30 minutter, fulgt av kombinasjonsbehandling med 10 mM HMBA i 24 timer. Proteinekstrakter ble immunoutfelt med et anti-CDK2 antistoff. Den CDK2 forbundet p27Kip1 i den resulterende immunkomplekser ble analysert lik A. D, SGC7901 celler ble infisert med Ad-HA-GSK-3βCA eller vektor kontroll (Ad-β-gal) ved en MOI på 10 PFU /celle. Etter 48 timers inkubering, ble hel-celle proteinet ekstraheres og immunopresipitert med anti-CDK2-antistoff (øvre panel). p27Kip1 ble analysert ved western blotting lik A. Overekspresjon av HA-merket GSK-3ca ble bekreftet ved Western-blotting ved anvendelse av anti-GSK-3β antistoff (lavere panel). GSK-3β aktivitet ble analysert ved en in vitro
kinase-analyse ved hjelp av sneglen protein som substrat (nederst panel).
Diskusjon
PI3-kinase /Akt /GSK-3β signalveien har vært innblandet i reguleringen av cellevekst, differensiering og apoptose av forskjellige celletyper [46]. I den foreliggende undersøkelse, hemming av GSK-3β ved hjelp av komplementære fremgangsmåter (dvs. kjemisk inhibering og konstitutivt aktiv Akt over-ekspresjon) attenuert protonpumpe-ekspresjon, en måling av mavelignende differensiering, i den gastriske tumor-avledede SGC7901 cellelinje.
celleproliferasjon og differensiering er tradisjonelt oppleves som gjensidige prosesser, med cellesyklus uttak er nødvendig for terminal differensiering [40], og P27 Kip1 spiller en viktig rolle [41, 42]. Genetisk sletting av p27, men ikke p21 er blitt vist å påvirke gastrisk celledifferensiering, mens tvunget p27 Kip1 ekspresjon fører til differensiering, noe som tyder på at p27 Kip1 er viktigere enn p21 Waf1 i regulering av gastrisk celle Differensiering. I overensstemmelse med disse funn hemming av GSK-3β svekkede HMBA-mediert gastrisk celle differensiering og hemming av GSK-3β blokkert HMBA-mediert atom p27 Kip1 uttrykk, mens overekspresjon av den aktive formen av GSK-3β øket kjerne p27 Kip1 uttrykk, noe som tyder på en viktig rolle i reguleringen av mage celledifferensiering gjennom regulering av atom p27 Kip1 uttrykk.
Other Languages
- fr:P27kip1, régulée par la glycogène synthase kinase 3β, aboutit à une différenciation induite par le HMBA des cellules cancéreuses gastriques humaines
- de:P27KIP1, von Glykogen-Synthase-Kinase-3β reguliert wird, führt zu HMBA-induzierte Differenzierung von humanen Magenkrebs cells
- es:P27KIP1, regulado por la glucógeno sintasa quinasa-3β, da como resultado la diferenciación inducida por HMBA de P27 cells
- nl:P27kip1, gereguleerd door glycogeen synthase kinase-3β resulteert in HMBA-geïnduceerde differentiatie van menselijke maagkanker cellen
- da:P27KIP1, reguleret af glycogensyntasekinase-3β resulterer i HMBA-induceret differentiering af humane gastriske cancerceller
- sv:P27Kip1, regleras av glykogensyntaskinas-3β, resulterar i HMBA-inducerad differentiering av humana gastriska cancerceller
- fi:P27Kip1, säätelee glykogeenisyntaasikinaasi-3β, tulokset HMBA aiheuttama erilaistumista ihmisen mahasyövän cells
- no:P27Kip1, regulert av glykogen syntase kinase-3β, resulterer i HMBA-indusert differensiering av humane magecancerceller
- sk:P27Kip1, regulované kináza glykogénsyntázu-3p, vedie k diferenciácii HMBA indukovanej ľudských buniek karcinómu žalúdka
- it:P27Kip1, regolato da glicogeno sintasi chinasi-3β, si traduce in differenziazione HMBA-indotta delle cellule di cancro gastrico umano
- pt:P27KIP1, regulamentada pelo glicogênio sintase-quinase-3β, resulta na diferenciação induzida HMBA de P27 cells
- sl:P27Kip1, ureja glikogen sintazo kinazo-3p, rezultati v HMBA-inducirane diferenciacijo človeške želodčne raka cells
- lt:P27Kip1, reglamentuoja glikogensintazę kinazės-3β, rezultatai HMBA sukeltos diferenciacijos žmogaus skrandžio vėžys cells
- hu:P27Kip1, szabályozza a glikogén szintáz kináz-3β, az eredmények HMBA által kiváltott differenciálódását emberi gyomor rákos sejtek
- ru:P27Kip1, регулируется гликоген-синтазы-киназы-3 &beta приводит к ГММК-индуцированной дифференцировки клеток рака желудка человека
- lb:P27Kip1, vun Glucogène synthase kinase-3β reegelen, Resultater am HMBA-entschlof dat vu mënschlechen gastric Kriibs cells
- hr:P27Kip1, regulirani glikogen sintetaze kinaze-3β, rezultira HMBA-inducirana diferencijacije humanog želučanog raka cells
 Lungemikrober kan bidra til å forutsi utfall hos alvorlig syke
Lungemikrober kan bidra til å forutsi utfall hos alvorlig syke
 Sopp og bakterier i tarmen kan også påvirke menneskers helse og alvorlighetsgraden av sykdommen
Sopp og bakterier i tarmen kan også påvirke menneskers helse og alvorlighetsgraden av sykdommen
 Immunceller reparerer skadet tarm hos barn med IBD
Immunceller reparerer skadet tarm hos barn med IBD
 Utett tarm og mikrobiell dysbiose kan bidra til cytokinstorm i alvorlig syke COVID-19-tilfeller
Utett tarm og mikrobiell dysbiose kan bidra til cytokinstorm i alvorlig syke COVID-19-tilfeller
 Fordøyelsesmanifestasjoner er vanlige, men milde blant sykehusinnlagte COVID-19 pasienter
Fordøyelsesmanifestasjoner er vanlige, men milde blant sykehusinnlagte COVID-19 pasienter
 Forskere finner en ny måte å beskytte mot sykdom i MS -modellen
Forskere finner en ny måte å beskytte mot sykdom i MS -modellen
 Sædmikrobiom avslørt med RNA -sekvensering
En ny studie rapporterer den første noensinne detaljerte beskrivelsen av det menneskelige sædmikrobiomet, ved bruk av nyere RNA -sekvenseringsteknikker som er i stand til å skille mellom sæd -RNA og b
Sædmikrobiom avslørt med RNA -sekvensering
En ny studie rapporterer den første noensinne detaljerte beskrivelsen av det menneskelige sædmikrobiomet, ved bruk av nyere RNA -sekvenseringsteknikker som er i stand til å skille mellom sæd -RNA og b
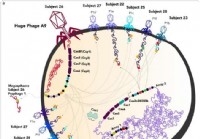 Nyoppdagede store fag utvisker grensen mellom liv og skade
En ny studie publisert i tidsskriftet Natur viser at det er bokstavelig talt hundrevis av virus som er store nok til å konsumere bakterier, og med egenskaper som er typiske for en levende organisme
Nyoppdagede store fag utvisker grensen mellom liv og skade
En ny studie publisert i tidsskriftet Natur viser at det er bokstavelig talt hundrevis av virus som er store nok til å konsumere bakterier, og med egenskaper som er typiske for en levende organisme
 Ulcerøs kolitt og en manglende mikrobe i tarmen
Ulcerøs kolitt er en alvorlig svekkende inflammatorisk sykdom i tarmen som fører til lammende symptomer som kan påvirke livskvaliteten alvorlig. Forskere fra Stanford University School of Medicine har
Ulcerøs kolitt og en manglende mikrobe i tarmen
Ulcerøs kolitt er en alvorlig svekkende inflammatorisk sykdom i tarmen som fører til lammende symptomer som kan påvirke livskvaliteten alvorlig. Forskere fra Stanford University School of Medicine har