Samtidig kompositt adrenal pheochromocytoma, flere mage stromal tumor og pseudohermaphrodism hos en pasient med von Recklinghausen sykdom
Abstract
Selv feokromocytom forekommer hos 1% av pasienter med von Recklinghausen sykdom, kompositt svulster i dette syndromet er mye sjeldnere, med isolert tilfelle rapporter i litteraturen. De fleste gastrointestinal stromal tumor (GIST) er enslig og sporadisk. Flere GIST er imidlertid forbundet med kliniske syndromer særlig von Recklinghausen sykdom. Vi tror dette er den første rapporten av kompositt adrenal pheochromocytoma og flere GIST forekommer i en 82 år gammel kvinne med nevrofibromatose type 1 (NF1), manifestert ved klitoris og subkutane nevrofibromer, epilepsi og Lisch knuter. Den ekstreme forstørret klitoris reist bekymringer av pseudohermaphrodism på presentasjonen
Innledning
Nevrofibromatose type 1 (NF1) er en heterogen, dominant arvelig lidelse forbundet med et stort antall forskjellige svulster, både godartede og ondartede.. Feokromocytom forekommer hos 1% av pasientene med NF1, mens kompositt svulster i binyremargen er mye sjeldnere. Per definisjon er et sammensatt svulst består av pheochromocytoma med neuroblastom, ganglioneuroblastoma eller ganglioneuroma. Begge cellelinjene, kromaffinceller og symapathetic gangliecellene er av neural crest opprinnelse. Gastrointestinal stromal tumor (GIST) er de vanligste mesenkymale svulster i mage-tarmkanalen, og har også blitt beskrevet i forbindelse med NF1. GIST er immunhistokjemisk positiv for KIT tyrosin kinase reseptoren og denne egenskapen er også funnet i de interstitielle celler av Cajal, de pacemakercellene regulerer autonom motorisk aktivitet. Det er postulert at GIST stammer fra en multipotential forløper celler, som skiller en interstitiell celle Cajal fenotype. Nevrofibromer av klitoris er rapportert sjelden. Det representerer en differensialdiagnose som må vurderes i pseudohermaphrodism.
NF1 har vært forbundet, om enn sjelden, med kompositt ganglioneuroma-ganglioneuroma av binyremargen, multifokale mage GIST og neurofibroma av klitoris, men vi tror dette er den første kasuistikk der alle disse sykdommene forekommer i samme pasient med NF1.
case report
en 82 år gammel enke og mor til tre med nevrofibromatose ble innlagt på sykehus etter et anfall på grunn av manglende overholdelse av krampestillende medisiner Hennes epilepsi hadde tidligere er godt kontrollert på fenytoin 300 mg daglig. Kranie computertomografi (CT) viste en gammel venstre frontal cerebral infarkt. Pasientens andre tidligere historie inkludert mild aortastenose, brukket venstre halsen på femur og jernmangelanemi. Gastroskopi og koloskopi ble utført 3 år tidligere, da hun ble undersøkt for melena og ingen åpenbar årsak funnet. Glibenklamidbehandlede 5 mg tds hadde blitt startet av hennes fastlege for symptoma glycosuria. Hun var på ingen andre medisiner.
Løpet av denne adgang, pasienten utviklet en gastrointestinal psevdoobstruksjon, som ble administrert konservativt. Det var under etterforskningen av hennes forstoppelse og progressiv oppblåst mage som forstørret klitoris ble notert på undersøkelse og en 2,5 cm venstre binyre masse funnet på abdominal CT.
Pasienten ga en historie med klitoris utvidelse fra fødsel og minnes klitoris reduksjon kirurgi blir tilbudt og gikk ned på 4-års alder. Hun utviklet en rekke subkutane knuter på 16-år og kutan neurofibroma ble bekreftet på biopsi av knuter i huden på den tiden. Det var en familiehistorie med nevrofibromatose, som rammet både sin mormor og oldemor, og pasientens egen yngste datter.
Gitt sammenslutning av nevrofibromatose og pheochromocytoma, ble pasienten spørres om paroksysmal symptomer på hjertebank, hodepine, svette , rødme eller synsforstyrrelser, men disse var fraværende. Ved undersøkelse var hun normotensive med en postural nedgang fra 135/60 mmHg til 110/60 mmHg. Puls var 80 og regelmessig, og det var en hørbar, myk, aortic stenotisk mumling. Utbredt kutane nevrofibromer og café-au-lait flekker ble notert. Forstørret klitoris var til stede med klitoris måler 10 × 4 × 4 cm. De visuelle felt og fundi var normale selv Lisch knuter var tilstede til venstre iris. Pasienten hadde bilateral sensineural hørselstap, men ingen andre nevrologiske underskudd ble oppdaget
Biokjemiske undersøkelser bekreftet forhøyet 24-timers urin catecholamine utskillelse 6 dager etter innleggelse. Adrenalin 232 nmol /dag (referanseintervall < 120 nmol /dag), noradrenalin 465 nmol /dag (ref. < 600); og 24-timers urin kreatinin 4,6 mmol /dag (ref. 5,0 til 16,0). En gjenta 24-timers urin catecholamine tre dager senere også bekreftet vedvarende heving av urin adrenalin av 149 nmol /dag med urin kreatinin på 4,4 mmol /dag. Urinutskillelse av kortisol var 530 nmol /dag (ref. ≪ 590). Pasienten var biokjemisk euthyreoide med gratis tyroksin av 13 pmol /L (ref. 9-19) og TSH 3,15 mU /L (ref. 0,30 til 4,00). Prolaktin var 380 mU /L (ref. ≪ 420), og hun ble normocalcemic, total plasma kalsium korrigert til albumin 40 g /L var 2,47 mmol /L og ionisert kalsium 1,18 mmol /L (ref (ref 2,25 til 2,60.) . 1,12-1,32) justert til pH på 7,4. Serumet kjønnshormonet profilen avslørte testosteron 3,1 nmol /L (ref. ≪ 3,5), østradiol 110 pmol /L (ref 40-130.), Gratis androgen index 2.3 (ref 1-5.), DHEA-sulfat < 0,2 umol /L (ref. 0,4 til 2,4), og androstendion 4,4 nmol /L (ref. 01.06 til 09.03). Hennes serum 17-hydroxy progesteron ble mildt forhøyet i 2,3 nmol /L (ref. 0,3 til 1,8), men dette ble ansett å være uten klinisk betydning. Cytogenetisk evaluering av perifert blod viste en normal kvinnelig karyotype.
Gitt vedvarende forhøyet urin adrenalin utskillelse, CT funn av en 2,5 cm igjen adrenal svulst var sannsynlig å være funksjonell og dette ble bekreftet ved hjelp av
123I-MetaIodoBenzyl Guanidine ( MIBG) scintigrafi. Pasienten ble klargjort for operasjon med fenoksyben 30 mg bd en uke før operasjon med propranolol 40 mg bd lagt dagen før operasjonen. Laparoskopisk venstre adrenalektomi ble foretatt, men dette ble omgjort til en åpen adrenalektomi grunn av tekniske problemer. I løpet av denne prosedyren ble omtrent ti knuter som måler mellom 3 og 5 mm bemerket på den serosale overflate av magen, hvorav den ene ble vevsprøve. Inspeksjon av resten av intra-abdominal viscera var dagligdags. Kjerne biopsier av forstørret klitoris ble tatt. Den postoperative kurset var begivenhetsløs og pasienten gjort en utmerket utvinning. Gjenta urin catecholamine utskillelse før lossingen var normal.
Ved to-måneders oppfølging etter henne adrenalektomi, en clitorectomy ble utført når en smertefull sår utviklet på klitoris.
Patologiske funn
Gastric serøse nodule
Makroskopisk biopsi besto av en uregelmessig stykke fast rosa tan vev måler 6 × 5 × 4 mm. Mikroskopisk var det en Lobulær svulst som består av langstrakte spindel ensartede celler adskilt av kollagene stroma. Kjernene ble arrangert i Palisades, og det var ingen cytologiske atypia og ingen mitoser var til stede. Små foci av tumor forlenget inn i det tilstøtende glatt muskulatur. Immunhistokjemi viste sterk og diffus farging for c-kit (CD117) og CD34. Det var også fokal farging for desmin og glatt muskulatur aktin. Det var ingen farging med S100. Funnene var konsistente med en liten gastrointestinal stromal tumor (GIST).
Klitoris biopsi og clitorectomy
biopsi besto av en kjerne av myk, hvitaktig grå vev måler 9-x-7x-1,5 mm og hele svulsten bestod av en hud dekket polypoid masse måler ca 90 x 40 mm. Det var en stor nyreformede sårdannelse med godt definerte hvite marginer som måler 43 x 30 mm. Mikroskopisk var det små avgrensede reir av nevrale typen strukturer som minner om sensoriske Meissnerian mene atskilt med litt myxoid stroma inneholder mange mastceller. Fokalt, var det en diffus arkitektonisk mønster av dårlig definerte bunter av cytologisk ensartede spindel celler med rikelig fibrillary matrise. Immunhistokjemi viste S100 positivitet. Histologisk diagnose var en klitoris neurofibroma.
Venstre adrenal svulst
venstre binyrene målt 48 x 25 x 50 mm og veide 11 gram. Svulmende over overflaten var en nodule måler 20 × 20 × 13 mm, som var spraglete grå-gul-orange på snittflaten. En 6 mm gråaktig nodul ble funnet i den tilstøtende fettvev. Mikroskopisk, de fleste av svulsten (70%) viste typiske trekk ved en pheochromocytoma, med diskrete reder av kreftceller separert av delikat fibro-vaskulær septa. Disse cellene var av stor med iøynefallende granulær cytoplasma og viste pleomorphism, mange celler som har forskjellige nucleoli. Mitotiske tallene ble ikke sett, og det var ingen nekrose. Cellene utvidet gjennom kapselen fokalt inn i peri-binyre fett. Ingen vaskulær invasjon ble sett. I noen få områder svulstcellene ble mer allment atskilt med tykke bånd av kollagen. Den andre komponenten av tumoren (30%) viste trekk ved ganglioneuroma, med store, modne ganglion-type celler med store kjerner, sentral eosinofil nucleoli og rikelig cytoplasma innleiret i et fibrillært neurale nettvare inneholdende cytologisk ensartede spindelformede celler. Ingen primitive nevroblastiske elementene ble sett. De to kreft komponenter ble godt avgrenset på noen områder, men fokalt dukket blandet.
Immunohistochemistry viste ingen farging av tumor elementer med spredning markør Ki-67 eller p53 protein. Positiv farging for S100 protein ble identifisert i sustentacular celler i løpet av normale adrenal medulla og i phechromocytoma, så vel som i gangliecellene til ganglioneuroma. Adrenal svulst var derfor en blandet pheochromocytoma /ganglioneuroma eller kompositt feokromocytom.
Nodule bemerket ved siden av hoved adrenal hevelse besto av en forstørret ganglion inkludert utvidet og forvrengt nervefibre. Skinn foreslo fokus involvering av neurofibroma.
Diskusjon
Nevrofibromatose type 1 (NF1) er en av de vanligste dominant arvet lidelser, med en fødsels forekomst av en i 3000 [1]. Den klassiske beskrivelsen gitt av patolog von Recklinghausen mer enn et århundre siden samsvarer godt med de mest vanlige kliniske bildet [2]. National Institutes of Health Consensus Statement [3] skissert syv diagnostiske kriterier for NF1 og to eller flere kriterier må være til stede for å stille en diagnose. Disse kriteriene omfatter: 6 eller flere café-au-lait macules (størrelse > 5 mm i pre-pubertale pasienter og størrelse > 15 mm i post-pubertale pasienter); 2 eller flere nevrofibromer eller 1 eller flere plexiform nevrofibromer; armhulen eller lyske freckling; 2 eller flere Lisch knuter; optiske hjernesvulst; spesifikke ossøs dysplastiske lesjoner; eller en første-graders slektning med NF1 diagnostisert med kriteriene ovenfor.
Expression of NF1 er svært variabel. Pasienter med denne lidelsen har en økt forekomst av både godartede og ondartede svulster. Dette ble vist i en undersøkelse av 70 voksne NF1 pasienter i Sverige [1]. Ondartede svulster ble rapportert fire ganger så ofte i NF1 gruppen enn i befolkningen generelt. Av den NF1 gruppen, hadde 24% utviklet maligne tumorer (16% karsinom, sarkom 7%, 1% melanoma), og 17% hadde utviklet godartede svulster (7% GIST, 6% pheochromocytoma, 3% adenom og 1% meningioma). Feokromocytomer (Pheo) er godt anerkjent i innstillingen av NF1 og sies å være 10 ganger mer vanlig enn den generelle befolkningen [4]. NF1 er funnet i 5-25% av pasientene med Pheo [4], mens den totale forekomsten av Pheo i NF1 er 1% [5].
Kompositt binyremargceller tumorer, på den annen side, er meget sjeldne. I en analyse fra National Cancer Institute of 120 binyrene og ekstra adrenal Pheos, bare fire hadde et sammensatt fenotype, noe som gjør en total forekomst av 3% [7]. En gjennomgang av Moore og Biggs [8] fant bare 13 tilfeller av kompositt pheochromocytoma-ganglioneuroma (pheo-GN) rapportert i litteraturen. Av disse bare tre rapporter funnet kompositt Pheo-GN forekommer i innstillingen av NF1 [6, 9, 11]. Vår sak rapport representerer den fjerde slik rapportert forekomst. I tillegg har en adrenal ganglioneuroma og kontralateral Pheo hos en pasient med NF1 blitt rapportert [10].
Bolande [12] var den første til å foreslå begrepet neurocristopathies eller svulster som stammer fra neural crest-deriverte celler. Disse svulstene av neural crest opprinnelse er sammensatt av ulike kombinasjoner av Binyremarghyperfunksjon eller ekstra adrenalin kromaffinceller, neuroblasts eller sympatisk ganglion celler av varierende stadier av differensiering, enten godartet eller ondartet. Pheo, paragangliom, GN, neuroblastom og neurofibroma alle representerer slike neurocristopathies.
Sympathogonia avledet fra visse neural crest cellene differensieres til neuroblasts og pheochromoblasts. Disse neural crest-avledede celler migrerer ventralt og differensiere for å gi opphav til de cellulære komponenter av det sympatiske ganglier og adrenal medulla. Enhver forstyrrelse eller migrering maldevelopment av den nevrale kløft kan resultere i utvikling av kompositt tumorer. Ettersom disse cellepopulasjoner har lignende utledning man kunne forvente kompositt binyremargceller tumorer for å være mer vanlig enn det som fremgår av litteraturen, og dette kan representere underrapportering eller under-gjenkjennelse av denne tilstanden.
NF1 påvirker den gastrointestinale traktus i 10 -25% av tilfellene [15] og det gjør det i fire prinsipielle former: stromal tumor, nevronale hyperplasi, ganglioneuromatosis og endokrine svulster i tolvfingertarmen og periampullary regioner. Akutt eller tilbakevendende gastrointestinal blødning kan forekomme hos opptil 40% av disse pasientene [16]. Gastrointestinal stromal tumor (GIST) er mesenkymale tumorer i mage- og tarmkanalen, som ikke har noen av de identifiserende egenskaper som indikerer en opprinnelse fra en spesifikk bindevev celle. De skiller seg fra glatte muskelceller svulster både immunophenotypically og molekylært. Nylig ultra likheter er funnet mellom GIST celler og interstitiell celle i Cajal (en gastrointestinal pacemaker celler) [13] og dessuten er det immunhistokjemiske likhetstrekk med både uttrykker KIT tyrosin kinase reseptoren. Det har blitt foreslått at GIST kan stamme fra stamceller som skiller mot en pacemaker celle og glattmuskelcelle. GIST kan uttrykke et antall forskjellige fenotyper, noe som har resultert i en rekke akronymer som brukes for å beskrive dem [14]. Myoid celletyper har blitt kalt glatte muskelceller GIST (smGIST), nervecelletyper kalt gastrointestinale autonome nervesvulster (GANT), Cajal celletyper kalt gastrointestinale svulster pacemaker celler (GIPACT) og blandede celletyper (blandet GIST). Den biologiske oppførsel av GIST har vært vanskelig å forutsi. Multivariat analyse av overlevelsesdata i 1004 GIST har vist tumor størrelse og plassering, mitotisk hastighet og alder til å være viktige prognostiske faktorer [17]. Imidlertid har ingen metode eller kombinasjon av metoder vist seg å være pålitelig i å bestemme malignitet i alle tilfeller.
Urin nevrofibromer er sjeldne og klitoris engasjement i NF1 har blitt rapportert sjelden. Men når det skjer, er forstørret klitoris ofte presentere tegn. I mange tilfeller er det medfødt. I en studie av 236 familier med NF1, fire pasienter hadde klitoris engasjement og i tre, ble involvering begrenset til klitoris [18]. En gjennomgang av litteraturen ved Sulphen [18] fant 26 pasienter med NF1 og klitoris engasjement.
Denne saken rapporten illustrerer heterogenitet NF1 med samtidig presentasjon av tre sjeldne manifestasjoner av denne fascinerende lidelse, nemlig kompositt pheo-GN av binyremargen, flere mage GIST og neurofibroma av klitoris. Så vidt vi vet er dette første gang en slik sak har blitt rapportert i litteraturen. Figur 1 A: Neurofibroma av klitoris. . B: forstørret klitoris
Figur 2 Gastric gastrointestinal stromal tumor (GIST) som viser fascicles av ensartede spindel celler (A, H & E opprinnelige forstørrelse × 250) og med c-kit farging av tumorceller sammenlignet med fraværende flekker i tilstøtende glatt muskulatur . oppe til venstre (B, c-kit opprinnelige forstørrelse x 100)
Figur 3 Neurofibroma av klitoris som viser en vekst mønster som ligner Meissner organer (H & E, opprinnelig forstørrelse × 250)
Figur 4 Composite adrenal svulst med. feokromocytom (venstre nedre) sammenslåing med ganglioneuroma (øverst til høyre). (H & E, opprinnelig forstørrelse × 250)
Erklæringer
Forfattere 'originale legges filer for Images Nedenfor er linkene til forfatternes opprinnelige innsendte filer for bilder. 12956_2006_42_MOESM1_ESM.pdf Forfatteroriginalfilen for figur 1 12956_2006_42_MOESM2_ESM.tiff Forfatteroriginalfilen for figur 2 12956_2006_42_MOESM3_ESM.tiff Forfatteroriginalfilen for figur 3 12956_2006_42_MOESM4_ESM.tiff Forfatteroriginalfilen for figur 4
 Strategisering av beredskap for pediatrisk helse for den andre bølgen av COVID-19-pandemien
Strategisering av beredskap for pediatrisk helse for den andre bølgen av COVID-19-pandemien
 Immunceller reparerer skadet tarm hos barn med IBD
Immunceller reparerer skadet tarm hos barn med IBD
 Forskere utvikler 3D -trykt pille som prøver bakterier som finnes i tarmen
Forskere utvikler 3D -trykt pille som prøver bakterier som finnes i tarmen
 Hvite blodlegemer og deres rolle i hjernen
Hvite blodlegemer og deres rolle i hjernen
 Infliximab kan hindre effektiviteten av noen COVID-19-vaksiner
Infliximab kan hindre effektiviteten av noen COVID-19-vaksiner
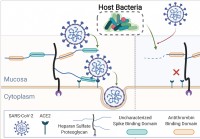 Menneskelig mikrobiom beskjærer slimhinneglykaner,
Menneskelig mikrobiom beskjærer slimhinneglykaner,
 Nyfødt musemodell gir ledetråder til årsaken til ødeleggende tarmsykdom i anemiske preemier
Leger har lenge mistenkt at transfusjon av røde blodlegemer gitt til premature spedbarn med anemi kan sette dem i fare for å utvikle nekrotiserende enterokolitt, eller NEC, en potensielt dødelig infla
Nyfødt musemodell gir ledetråder til årsaken til ødeleggende tarmsykdom i anemiske preemier
Leger har lenge mistenkt at transfusjon av røde blodlegemer gitt til premature spedbarn med anemi kan sette dem i fare for å utvikle nekrotiserende enterokolitt, eller NEC, en potensielt dødelig infla
 Mikrober kan forutsi dødelige utfall hos ventilerte COVID-19-pasienter
Nærværet av Mycoplasma salivarium i de nedre luftveiene til ventilerte pasienter med COVID-19-infeksjon er assosiert med økte sjanser for å dø. Resultatet var en del av en molekylær undersøkelse som
Mikrober kan forutsi dødelige utfall hos ventilerte COVID-19-pasienter
Nærværet av Mycoplasma salivarium i de nedre luftveiene til ventilerte pasienter med COVID-19-infeksjon er assosiert med økte sjanser for å dø. Resultatet var en del av en molekylær undersøkelse som
 Ny forskning identifiserer en kobling mellom tarmmikrobiomet og slag
En nylig studie utført på Cleveland Clinic demonstrerer virkningen av dietter som inneholder mye kolin, som ofte finnes i store mengder i rødt kjøtt og eggeplommer, og trimetylamin om økende slaglengd
Ny forskning identifiserer en kobling mellom tarmmikrobiomet og slag
En nylig studie utført på Cleveland Clinic demonstrerer virkningen av dietter som inneholder mye kolin, som ofte finnes i store mengder i rødt kjøtt og eggeplommer, og trimetylamin om økende slaglengd