en ny metode, digital genomet skanning oppdager KRAS
genamplifisering i mage kreft: involvering av overexpressed villtype KRAS i nedstrøms signal og kreft celle vekst
Abstract
Bakgrunn
Magekreft kreft~~POS=HEADCOMP er den tredje vanligste kreftformen som påvirker den generelle befolkningen over hele verden. Avvikende aktivering av KRAS er en nøkkelfaktor i utviklingen av mange typer svulst, men onkogene mutasjoner av KRAS
er sjeldne i magekreft. Vi har utviklet en ny metode for kvantitativ analyse av DNA-kopi antall, betegnet digitalt genomet skanning (DGS), som er basert på bestemmelse av korte restriksjonsfragmenter, og ikke involverer PCR eller hybridisering. I denne studien har vi brukt DGS å kartlegge kopi-nummer endringer i mage kreftceller.
Metoder
DGS av magekreft cellelinjer ble utført ved hjelp av sekvenser av 5000 til 15000 restriksjonsfragmenter. Vi screenet 20 mage kreft cellelinjer og 86 primær mage svulster for KRAS
forsterkning av kvantitativ PCR, og undersøkt KRAS
forsterkning på DNA, mRNA og proteinnivåer av mutasjonsanalyse, real-time PCR, immunoblot analyse, GTP -RAS pull-down analysen og immunhistokjemisk analyse. Effekten av KRAS
knock-ned på aktivering av P44 /42 MAP kinase og AKT og på cellevekst ble undersøkt av immunoblot og kolo analysen, henholdsvis.
Resultater
DGS analyse av HSC45 magekreftcelle linje viste amplifikasjon av en 500-kb region på kromosom 12p12.1, som inneholder KRAS
gen locus. Forsterkning av KRAS
locus ble påvist i 15% (3/20) av magekreft cellelinjer (8-18 ganger forsterkning) og 4,7% (4/86) av primære mage svulster (8-50 ganger forsterkning ). KRAS
mutasjoner ble identifisert i to av de tre cellelinjene som KRAS
ble amplifisert, men ble ikke påvist i noen av de primære tumorer. Overekspresjon av KRAS protein direkte korrelert med økt KRAS
kopiantall. Nivået av GTP-bundet KRAS ble forhøyet følgende serum stimulering i celler med forsterket villtype KRAS
, men ikke i celler med forsterket mutant KRAS
. Knock-down av KRAS
i mage kreftceller som gjennomføres forsterket villtype KRAS
resulterte i hemming av cellevekst og undertrykkelse av P44 /42 MAP kinase og AKT aktivitet.
Konklusjon
Vår studie fremhever nytten av DGS for identifisering av kopi-nummer endringer. Ved hjelp av DGS identifiserte vi KRAS
som et gen som forsterkes i human magekreft. Vi viste at genamplifikasjon sannsynlig danner molekylære grunnlaget for overaktivering av KRAS i magekreft. Ytterligere studier ved hjelp av en større kohort av magekreft eksemplarer er nødvendig for å fastslå de diagnostiske og terapeutiske implikasjoner av KRAS
forsterkning og overekspresjon.
Bakgrunn
Magekreft er den tredje vanligste kreftformen som påvirker den generelle befolkningen over hele verden [1 ]. Spesifikke genetiske endringer har blitt rapportert i magekreft, inkludert de presiseringer av KSAM
, MET Hotell og ErbB2
, og mutasjoner i p53
, APC
, og CDH1 product: [2] . Mens gain-of-funksjon mutasjoner av KRAS
er noen av de mest observerte genetiske endringer i en rekke svulster, inkludert bukspyttkjertelen (60%), galleveier (33%) og tykktarms (32%) [3], disse mutasjonene er sjeldne i magekreft (2-7%) [4-7]. Generelt RAS
mutasjoner assosiert med tumorigenesis "låse" RAS i en aktiv GTP-bundet tilstand. GTP-RAS binder seg til en rekke av effektor-proteiner for å stimulere nedstrøms signalbaner, hvorav de RAF-MAP-kinase-kaskaden og fosfatidylinositol 3-kinase (PI3K) -AKT veier for cellevekst og onkogenese er den best karakteriserte [3]. Langvarig aktivering av RAS kan også forekomme gjennom mekanismer som ikke involverer mutasjoner i RAS. For eksempel redusert uttrykk for la-7 microRNAs, som undertrykker RAS ved å målrette den 3'untranslated regionen RAS
mRNA, er ofte forbundet med en høyere RAS protein nivå i tumorer [8]. Til dags dato, de molekylære mekanismer for onkogen aktivering av RAS i magekreft er ikke fullstendig klarlagt.
Amplifisering av genomiske sekvenser som inneholder gener som er kritisk for cellevekst er en av de viktigste mekanismer for aktivering av onkogener i kreft, og er ofte forbundet med tumorprogresjon, dårlig prognose og /eller medikamentresistens [9]. Av de mange metoder som er tilgjengelige for å detektere kopinummer endringer genom-wide, er dagens gullstandard array CGH-metoden (aCGH). I løpet av de siste få årene, har oppløsningen på aCGH forbedret hurtig ved bruk av oligonukleotid-prober, og har overgikk aCGH ved anvendelse av standard BAC-prober [10]. Imidlertid er aCGH også utsatt for den iboende støy av hybridiserings-baserte intensitetsmålinger, som signalkvaliteten påvirkes av repetitive sekvenser, og er avhengig av sonden kvalitet [11]. Faktisk har optimalisering av sonde utforming vært en stor utfordring i utviklingen av flislegging matriser [12, 13] ble Digital karyotypering (DK).
Utviklet av Wang et al. [14], og er ikke begrenset av de iboende problemer med matrise teknikker. DK omfatter den digitale telling av korte fragmenter av genomisk DNA (betegnet lapper), som gir et kvantitativt mål på DNA-kopi antall gjennom tetthet analyse tag langs hvert kromosom. DK har vært brukt med hell til en rekke krefttyper å oppdage kopi-nummer endringer, blant annet forsterkning av TYMS
, RSF1 Hotell og OTX2
, og sletting av MKK4 Hotell og dystrophin product: [ ,,,0],15-19]. Til tross for effektiviteten av DK, er det teknisk vanskelig for brede anvendelser, fordi det involverer PCR-amplifisering og generering av koder av 21-basepar (bp) lengde for nøyaktig å representere den kromosomet sted av interesse.
Vi rapporterer her utvikling av en ny fremgangsmåte, betegnet DGS, for den kvantitative analysen av kopiantall variant, som er basert på den merkelapp-telling begrepet DK, men benytter en forenklet fremgangsmåte for fremstilling tag. DGS av magekreft cellelinjer oppdaget forsterkning av KRAS
locus på kromosom 12p12.1. Våre resultater gir en molekylær basis for den overaktivering av KRAS, og antyder at aktiveringen av KRAS nedstrøms signaleringshendelser kan fremme magecancer celleproliferasjon.
Methods
cellelinjer og vev
Cellelinjene som ble analysert i den aktuelle studien er oppført i tilleggsfiler 1. HSC og SH101P4 cellelinjer ble etablert av Kazuyoshi Yanagihara [20]; alle andre ble innhentet fra American Type Culture Collection eller den japanske Innsamling av forsknings Bioressurser (Tokyo, Japan). Alle cellelinjer ble dyrket i de anbefalte media. For serum stimulering, ble cellene inkubert i medium som manglet serum i 24 timer (h), og deretter enten ustimulert eller stimulert i 1 time med media inneholdende 10% føtalt kalveserum (FCS). Primære magekreft prøver ble innhentet fra Kirurgisk avdeling, Keiyukai Sapporo Hospital, med informert samtykke fra den enkelte pasient. Genomisk DNA ble ekstrahert ved hjelp av fenol-kloroform-metoden, etterfulgt av RNase-behandling. Total RNA ble ekstrahert ved hjelp Trizol (Invitrogen, Carlsbad, CA, USA), i henhold til produsentens instruksjoner. Genomisk DNA fra normale perifert blod leukocytter (Biochain, Hayward, CA, USA) og total RNA fra normal mageslimhinnen fra friske individer (Biochain og Invitrogen) ble kjøpt. Primære mage kreft ble klassifisert ved hjelp clinicopathological funksjoner, som vist i tilleggsfiler 2, i henhold til pTNM klassifiseringsordning (5. utgave, 1997) [21] og Lauren klassifiseringssystem [22]. KRAS
-amplification status i henhold til alder ble sammenlignet med Student t-test; i henhold til klasse, Pt status, pN status og sykdom scenen med Mann-Whitney U test; og i henhold til kjønn, histologi og PM status med nøyaktig Fisher test. Alle testene ble 2-tailed, og en P
verdi av < 0,05 ble ansett som statistisk signifikant
. Digital-genomet skanning
I korthet ble 40 pg av genomisk DNA ble underkastet restriksjonsenzymbehandling ved hjelp av Mbo
I (Takara, Tokyo, Japan) og deretter separert ved elektroforese på en 3% NuSieve GTG agarose gel. Korte fragmenter (30-60 bp, betegnes reelle tags) ble elektroeluert, sammensatt og subklonet i Bam
HI-kuttet pBluescript II KS + (Stratagene, La Jolla, California) bruker Mighty Mix DNA ligering løsning (Takara). Escherichia coli DH10B
ble transformert med de rekombinante plasmider, ble transformantene slått sammen og plasmid-DNA ble renset for å generere det første biblioteket. Concatemers av virkelige tagger ble skåret ut av Spe
I /Pst
-fordøyelse av det første biblioteket, og fragmenter i området fra 140 til 800 bp ble elektroeluert, sammensatt og subklonet inn i pBluescript II KS + for å generere det andre biblioteket. Andre bibliotek plasmider inneholder concatemers av Spe
I /Pst
fragmentene ble sekvensert ved hjelp av en ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), i henhold til produsentens instruksjoner. Unike virkelige koder ble kartlagt til menneskelige kromosom sekvenser, og tag tetthet, definert som forholdet mellom virkelige koder til virtuelle tags enn å flytte vinduer, ble beregnet til å oppdage uregelmessigheter i DNA-innhold ved hjelp av terskelverdiene som er definert av DGS simuleringer. Tag stillinger og forholdstall tag tetthet ble visualisert ved hjelp av tilpassede spor og Genome Grafer fra University of California, Santa Cruz (UCSC) genom nettleser (mars 2006 fryse, hg18) [23-25]. De detaljerte protokoller for DGS, virtuell tag karakterisering og i silikoaluminofosfater
simuleringer er tilgjengelig igjen fil 3.
kvantitativ real-time PCR
Relativ DNA kopiantall ble bestemt av kvantitativ real-time PCR ved hjelp av en SYBR Grønn PCR Master Mix (Applied Biosystems) og ABI PRISM 7000 (Applied Biosystems). DNA-innhold per haploid genom ble normalisert til det av en repeterende element, Line-en, og beregnes av komparative CT (ΔΔCT) relativ kvantifisering metode ved hjelp av formelen 2
(Nt
- nline
) - ( xt
- Xline
), der N
t
er terskelen syklus nummer observert for en eksperimentell primer i normal leukocytter DNA, er N
linjen
terskelen syklus nummer observert for line-en primer i normal leukocytter DNA, er Xt
gjennomsnittlig terskelen syklus nummer observert for den eksperimentelle primer i kreftcelle DNA, og X
linjen
er gjennomsnittlig terskel syklus nummer observert for linje-en primer i kreftcelle-DNA [14]. Genomisk forsterkning ble definert som en som er større enn 4 ganger økning i DNA-innhold. Primersekvensene for hver locus er tilgjengelige i tilleggsfiler 4. allel andelen muterte KRAS
(G12V, GGT → GTT) ble bestemt ved å bruke en modifisert real-time PCR prosedyre ifølge Itabashi et al product: [26 ]. Den detaljerte protokollen er tilgjengelig i tilleggsfiler 3. cDNA ble fremstilt ved anvendelse av Superscript III revers transkriptase (RT, Invitrogen), og mRNA nivå av hvert gen ble bestemt ved sanntids-RT-PCR ved anvendelse av TaqMan Gene Expression Assay (Applied Biosystems) . Relative mRNA-nivåer ble beregnet ved den komparative CT metode under anvendelse av GAPDH
som en endogen kontroll. De primer /probe sett som brukes er vist i tilleggsfiler 5.
Fluorescence in situ
hybridisering (FISH)
BACS som inneholdt KRAS
locus (RP11-636P12) og kromosom 12q24.2 (RP11 -91M21) ble merket med henholdsvis Cy3 og Cy5, og deretter inkubert med sklier tilberedt med interfase og metafase kromosomer. Kjerner ble kontra farget med 4 ', 6-diamino-2-phenylindole (DAPI), og lysbilder ble analysert ved hjelp av en fluorescens mikroskop (Leica CW-4000).
Mutasjonsanalyse av KRAS Hotell og PIK3CA
Amplified genomiske fragmenter ble sekvensert enten direkte eller subklonet ved hjelp av TOPO TA-kloningssettet (Invitrogen) og deretter sekvensert. Minst ti kloner fra to uavhengige PCR-analyser per locus ble sekvensert ved hjelp av M13 forover og bakover primere (Invitrogen). Sekvensene til primerne anvendt for amplifikasjon av KRAS
(eksoner 1 og 2) og PIK3CA
(exon 9 og 20) er vist i tilleggsfiler 6.
Immunoblot-analyse
Cellene ble lysert i lyserings buffer inneholdende 20 mM Tris-HCl (pH 7,5) buffer, 150 mM NaCl, 1 mM EDTA, 1% Triton X, 10% glycerol, 10 mM NaF, 1 mM natrium-vanadat, 50 mM β-glycerofosfat, 1 mM phenylmethansulfonyl fluor , 1 mM ditiotreitol, og en protease inhibitor cocktail (Roche, Mannheim, Tyskland). Proteiner ble separert ved hjelp av SDS-PAGE og elektroblottet på en Immobilon-P-membran (Millipore, Billerica, MA, USA). Membranene ble analysert ved immunblot anvendelse av følgende antistoffer, som antydet: mus monoklonalt anti-KRAS, -NRAS, og -HRAS antistoffer (sc-30, sc-31, og sc-29, respektivt, Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-aktin antistoff (Millipore); kanin polyklonale anti-P44 /42 MAP kinase, -phosho-P44 /42 MAP kinase (Thr202 /Tyr204), -Akt og -phospho-Akt (Ser473) sera (Cell Signaling Technology, Danvers, MA, USA).
GTP-RAS pull-down analysen
aktivering av RAS ble oppdaget ved hjelp av en EZ-Detect Ras Activation Kit (Pierce, Rockford, IL, USA). I korthet, ble cellelysatet (500 ug) ble inkubert med immobilisert Raf1 Ras-bindende domene fusert til glutation-S-transferase (GST-Raf1-RBD). Bunnfallet ble vasket 3 ganger, og bundne proteiner ble eluert ved koking i 5 minutter (min). Proteinene ble gjenoppløst på en 12% polyakrylamidgel, overført til en Immobilon-P-membran, og utsatt for immunoblot-analyse ved anvendelse av anti-KRAS, -NRAS, eller -HRAS antistoffer.
RNA-interferens, En spesiallaget KRAS
siRNA (5'-AGAGUGCCUUGACGAUACAdTdT-3 '), rettet mot en region av KRAS Hotell som ikke er knyttet til kjente onkogene mutasjoner, ble syntetisert ved Dharmacon (Lafayette, Co, USA). sirnas rettet mot LRMP
, LYRM5 Hotell og CASC1
ble kjøpt fra Ambion (No.144181, 284911 og 147715). En universell ikke-målsøkende siRNA (uspesifikk kontroll VII, Dharmacon) ble anvendt som en negativ kontroll. I hvert forsøk ble 5 x 10 6-celler ble transfektert med 7,5 pl 20 pM siRNA ved elektroporering (Amaxa, Køln, Tyskland) ved anvendelse av Nucleofector kit V eller T, i henhold til produsentens instruksjoner.
Celleproliferasjonsanalyse
Etter transfeksjon med sirnas, magecancercellelinjer HSC45, MKN1, AGS og NUGC4 ble sådd i 96-brønns plater ved en tetthet på 8000 celler /100 ul i standard medium inneholdende 10% FCS. Celle nummer på 48, 72 og 96 timer etter transfeksjon ble bestemt indirekte ved kolo analyse ved bruk av Cell Counting Kit-8-løsning (Dojindo, Kumamoto, Japan). Analysen er basert på reduksjon av et tetrazoliumsalt ([2- (2-metoksy-nitrofenyl) -3- (4-nitrofenyl) -5- (2,4-disulfofenyl) -2-tetrazolium, mononatriumsalt], WST -8) og blir brukt som et mål på levende celler. Absorbansen for hver brønn ved 450 nm ble målt ved anvendelse av en mikroplateavleser (modell 680, Bio-Rad, Hercules, CA, USA).
Strømningscytometri
Strømningscytometri ble utført som beskrevet tidligere [27]. Kort sagt ble adherente og frittliggende cellene høstet, fiksert i 90% kald etanol, behandlet med RNase A (500 enheter /ml), og deretter farget med propidiumjodid (50 ug /ml). For hver prøve ble 30000 hendelser analysert ved hjelp av cellesyklus analyse plattform FlowJo program (Tre Star, Ashland, OR, USA).
Immunohistochemistry
Formalin fiksert, parafininnstøpte deler av mage svulster ble deparaffinized, hydrert , og deretter behandlet med peroksidase blokkeringsoppløsning (3% H 2o 2 i metanol). Snittene ble autoklavert ved 105 ° C i 10 minutter i target henting løsning (Dako, Glostrup, Danmark). Seksjoner ble inkubert med et muse-anti-KRAS antistoff. (1: 100 fortynning, Santa Cruz Biotechnology) i 1 time ved romtemperatur, og immunoreaktivitet ble detektert ved bruk av ENVISION-Plus reagenser (Dako)
Resultater
Digital genom skanning og karakterisering av virtuelle koder i silico
Digital genomet skanning (DGS) er en metode for å kvantifisere genkopitallet ved opplisting korte genomiske DNA-fragmenter (kalt reelle tags) som genereres eksperimentelt ved Mbo
jeg endonukleasefordøyelse (Figur 1a). For å eliminere de kompliserte trinn som inngår i fremstillingen tag, vi beregnings karakterisert v de korte DNA-fragmenter som er produsert ved enkelt restriksjonsenzymkutting med Mbo
I, som gjenkjenner 4-bp sekvensen GATC. I silico
fordøyelsen av det menneskelige genom ved Mbo
jeg produserte om lag 1,6 millioner restriksjonsfragmentene (kalt virtuelle tags) i størrelsesorden 20-130 bp (tilleggsfiler 7a). Nukleotidsekvensanalyse viste at ca 65% av de virtuelle koder som finnes repeterende sekvenser, som definert i den offentlige databasen gjenta elementer (tilleggsfiler 7a). Viktigere, sekvens passer til det menneskelige genom database viste at ca 85% av de virtuelle koder kartlagt unikt til presise kromosom steder (tilleggsfiler 7b, c). Selv om de virtuelle kodene inkluderer repeterende sekvenser i en del, ca 80% av repeterende koder viste seg å være unik. Den gjennomsnittlige avstanden mellom to unike virtuelle tags på 30 til 60 bp i lengde var 7,6 kb, median avstand var 4,5 kb og 97,8% av intervallene var kortere enn 30 kb (tilleggsfiler 7d). Lignende tag intervall kjennetegn ble observert for virtuelle koder området 70-100 bp (gjennomsnittlig avstand, 7,9 kb, median avstand, 4,8 kb, 97,4% var kortere enn 30 kb), og 100-130 bp (gjennomsnittlig avstand, 7,9 kb; median avstand, 4,9 kb,.. 97,4% var kortere enn 30 kb (tilleggsfiler 7e, f) Videre tettheten av unike virtuelle koder var nesten lik i hvert kromosom (tilleggsfiler 7g) Disse in silico
funn antydet at flertallet av kort Mbo
i tags ville være informativ for DGS. Figur 1 DGS og utarbeidelse av reelle koder. (a) Skjematisk oversikt over DGS. Fargede bokser representerer genomisk Mbo
i reelle koder. Se teksten for detaljer. (b og c) Fremstilling (b) og karakterisering (c) virkelige koder. Representative resultater ved bruk av genomisk DNA fra MKN1 magekreftceller er vist. (b) korte fragmenter av Mbo
i-spaltet genomisk DNA (30 til 60 bp) ble elektroeluert fra en agarosegel (i), sammenkjedede og subklonet. Resulterende rekombinante plasmider ble slått sammen for å generere den første biblioteket (ii). Lange concatemers (140-800 bp) ble kuttet ut fra 1ste bibliotek vektorer, elektroeluert (iii), sammenkjedede og subklonet. De resulterende rekombinante plasmider representerer 2. bibliotek kloner (iv). Antallet koder som finnes i hver klon er vist øverst i hvert kjørefelt. Inserts ble undersøkt med Xhol
I /Sac
-fordøyelse i paneler (ii) og (iv). *, vektor fragmenter; **, Spe
I /Pst
jeg fordøyelse av flerkloningssetet uten innsatsen. (C) Faktisk antall reelle koder fra andre bibliotek vises i histogrammet (til venstre), og deres egenskaper er oppsummert (til høyre).
DGS simulering i silico
evne DGS å oppdage genom -Bredt endringer er basert på genom-egenskaper, slik som kopitallet og størrelsen av endring, og det antall virkelige koder som oppnås fra sekvensanalyse. For å forutsi størrelsen av endring som kan på en pålitelig måte skal detekteres, gis et fast antall av beregnings samplede koder, anvendte vi Monte Carlo-simulering for å beregne en positiv prediktiv verdi (PPV), som er sannsynligheten for at en detektert forandring representerer en sann endring. For eksempel fant vi at en analyse av 5000-koder kan pålitelig oppdage en 10-fold forsterkning på 500 kb, en homozygot delesjon på 7,5 Mb, eller en enkelt kopi tap av en 30 Mb regionen, men kunne ikke oppdage en subchromosomal få mindre enn 30 Mb (Tilleggs fil 8). Både sensitivitet og spesifisitet for å detektere slike endringer var > 99% i tilfeller med høye PPVs (mer enn 90%)., Noe som indikerte at hverken var en begrensende faktor i denne analyse (data ikke vist)
Fremstilling av reelle koder fra humant genomisk DNA
for DGS av magekreft cellelinjer HSC45 og MKN1, vi forberedt biblioteker av reelle koder fra genomisk DNA, som vist i Figur 1a. Den Mbo
I-spaltet genomisk DNA ble størrelsesfraksjonert (30-60 bp) og underkastet concatemerization, fulgt av konstruksjonen av et andre bibliotek, som inneholdt omtrent 10 reelle lapper pr klon (Figur 1b). Nukleotidsekvensanalyse av de virkelige koder viste at 85,8% tilordnet unike posisjoner, noe som var i tråd med vår karakterisering av virtuelle tags (Figur 1c).
Amplifikasjoner på kromosom 12p i HSC45 magekreftceller
genome-wide tag tetthet profilen til HSC45 celler ble bestemt ved hjelp av en totalt 5,462 unike reelle koder. For å oppnå høy oppløsning og følsomhet med de eksperimentelle data, har vi brukt vindusstørrelser på 1000 og 2100 virtuelle lapper (ca. 2300 kb og 4 700 kb) for analyse av amplifikasjoner og delesjoner, respektivt. Forholdet Koden tettheten ble beregnet som summen av reelle koder dividert med gjennomsnittlig antall virkelige koder i samme størrelse vinduer i hele genomet, hvor normal tag tetthet ratio ble definert som 1,0. Vi identifiserte forskjellige subchromosomal regioner av økt brikketetthet på 8q24.21, 12p12.1 og 12p13.33, og redusert tag tetthet på 9p21.3 og den lange arm av kromosom 18 (figur 2, tilleggsfiler 9a-d). Regionene økt brikketetthet (12p12.1, 12p13.33 og 8q24.21) inneholdt KRAS
, CACNA1C plakater (kalsium kanal, spenningsavhengig, l type, alfa-1c underenhet) og MYC
loci, respektivt. Southern blot-analyse bekreftet at KRAS Hotell og MYC
ble forsterket i HSC45 celler (tilleggsfiler 9e). Hver kopitall forandring kvantifisert som bestemmes kvantitativt real-time PCR (qPCR) av genomisk DNA var bemerkelsesverdig lik den som er beregnet ved DGS når vindusstørrelsen for analyse merkelappen Tettheten ble tilpasset størrelsen av hver endring (Tilleggs fil 9a-d ). Disse resultatene tyder på at merkelappen tetthet analyse av DGS kan brukes til å utføre kopitallet analyse i hele det humane genom. Figur 2 Påvisning av økt kopiantall på kromosomer 8q og 12p av DGS i HSC45 magekreftceller. (A) En hel-genomet riss av forholdet mellom merkelappen tetthet (ved hjelp av et vindu på 1000 virtuelle koder) i HSC45 celler som bestemt ved DGS. Verdiene på y-aksen indikerer brette forandringer i merkelappen tetthet i forhold til det gjennomsnittlige tag tetthet av hele genomet, og representerer DNA-innhold per haploid genom, i vinduer. X-aksen står for kromosomantall. (B og c) Utvidet riss av forholdstall tag tetthet på kromosom 8 (b) og 12 (c). I hvert panel viser den øvre kurve som en hel-kromosom riss av forholdet mellom merkelappen tetthet (basert på et vindu av 1000 virtuelle koder). Den nederste grafen viser et utvidet syn på 8q24.21 og 12p12.1, der økt brikketetthet ble oppdaget ved hjelp av vinduer i 1000 og 500 virtuelle koder. Unike virkelige koder vises som svarte loddrette streker, og unike virtuelle koder er angitt i blått (60 bp eller kortere) eller lyseblå (mer enn 60 bp) barer i tett modus. Posisjonene til refseq gener, med noen skjøting isoformer utelatt, vises nederst i de nedre panelene.
Forsterkning av KRAS
i mage kreft cellelinjer
Analyse av 26 loci innenfor og umiddelbart flankerer kromosom 12p12. 1 i HSC45 celler ved qPCR vist at et område på ca. 500 kb, som omfattet KRAS
gen locus, ble amplifisert (8-ganger amplifikasjon, figur 3a). Genomisk qPCR screening oppdaget KRAS
forsterkning i to ekstra mage kreft cellelinjer, SH101P4 (18 ganger) og MKN1 (13 ganger) (figur 3a), mens vi ikke registrerer forsterkning på mer enn fire ganger i 17 andre mage kreft cellelinjer, eller i 10 tykktarmskreft og 11 bukspyttkjertelkreft cellelinjer (som er oppført i tilleggsfiler 1, data ikke vist). DGS også oppdaget forsterkning av KRAS
locus i MKN1 celler (tilleggsfiler 10). De nærliggende gener av KRAS
i minimal amplicon var LRMP plakater (lymfoid-begrenset membran protein), CASC1 plakater (kreft mottakelighet kandidat 1) og LYRM5 plakater (LYR motiv som inneholder 5). BCAT1
(forgrenet aminotransferase 1, cytosoliske) ble også amplifisert i SH101P4 og MKN1 celler, men ikke i HSC45 celler. Vi bekreftet at CACNA1C
ble forsterket i HSC45 celler, men ikke i de andre mage, tykktarm, eller bukspyttkjertelkreft cellelinjer ved hjelp av genomisk qPCR analyse (tilleggsfiler 9b, data ikke vist). Verken NRAS
, HRAS
eller BRAF
presiseringer ble påvist i de ovennevnte kreftcellelinjer av genomisk qPCR analyse (data ikke vist). Forsterkningen av KRAS
ble også bekreftet av tofarget FISH analyse, hvor KRAS
amplicon var tydelig som et homogent-farget regionen i HSC45, SH101P4 og MKN1 celler (figur 3b). Figur 3 Geneamplifikasjon av KRAS i mage kreftceller. (A) Kvantitativ genomisk PCR-analyse av KRAS
locus på 12p12.1 i HSC45 celler. Diskret presiseringer på 12p12.1 i to andre mage kreft cellelinjer ble også påvist (SH101P4 og MKN1). DNA-kopitall i forhold til normal diploid leukocytt-DNA ble plottet mot kromosomalt nukleotid-posisjon (i megabases). Posisjonene til refseq gener i de tilsvarende områder er vist i den nederste kartet. Minimumsforsterkning region som er felles for alle 3 gastrisk cancer cellelinjer er representert ved den oransje-farget linje. (B) metafase (til venstre) - og inter (til høyre) -FISH analyse av de forsterkede KRAS
locus i mage kreft cellelinjer. KRAS
-spesifikke probe er i gult, og kontroll sonde, spesifikk for den lange arm av kromosom 12, er i rødt. ble observert Tetraploidy i HSC45 og triploidy i SH101P4 og MKN1 celler. (C) Kvantitativ real-time RT-PCR analyse av KRAS
mRNA uttrykk i magekreftceller med 12p12.1 forsterkning. Expression analyse av gener (KRAS, LRMP, CASC1 Hotell og LYRM5
) ligger innenfor minimal fragment, og BCAT1
, som flankerer minimal fragment, ble utført ved hjelp av real-time RT-PCR. Expression nivåer ble normalisert til GAPDH
mRNA, og er avbildet som en farge gradient i forhold til normal mage. Den genamplifisering og mutasjon (kodon 12 eller 13) status av KRAS
for hver prøve er oppsummert i de riktige to kolonner. Fylte sirklene indikerer tilstedeværelse av forsterkning eller mutasjon av KRAS
, og åpne sirkler indikerer ingen forsterkning eller ingen mutasjon av KRAS
.
Sekvens analyse av KRAS plakater (tilleggsfiler 11a) viste at både HSC45 og SH101P4 cellene næret en mutasjon i kodon 12 som resulterte i en enkelt aminosyre substitusjon i KRAS (GGT → GTT, G12V), mens MKN1 celler manglet KRAS
mutasjoner. Tilstedeværelsen av KRAS
mutasjoner i AGS (G12D), SNU1 (G12D), DLD1 (G13D) og HCT116 (G13D) celler er blitt rapportert tidligere [28, 29]. Av de ti PCR-kloner av KRAS
fra HSC45 og SH101P4 celler som ble utsatt for mutasjonsanalyse, åtte og tre, henholdsvis, næret mutasjoner i kodon 12. Videre genomisk real-time PCR-analyse ved hjelp av sonder som var spesifikk for vill -type og mutante alleler KRAS
(Tilleggs fil 11b) også viste at HSC45 og SH101P4 celler inneholder forskjellige andeler av det mutante allelet (80% og 50%, respektivt). Samlet er disse resultatene indikerte at forsterkningen av en mutant KRAS
allel forekommer også i HSC45 og SH101P4 celler.
Vi neste undersøkt nivåene av KRAS
mRNA i KRAS
-amplified mage kreftceller ved kvantitativ real -time RT-PCR (QRT-PCR) (figur 3c). Nivåene av KRAS
mRNA korrelerte signifikant med KRAS
kopiantall. De nærliggende gener LYRM5 Kjøpe og CASC1
, som lokaliserte til minimal fragment, ble også uttrykt på et høyere nivå i celler med forsterkning i forhold til celler uten forsterkning (figur 3c). Interessant nok LRMP
ble nedregulert i kreftceller sammenlignet med normale magen celler. Immunoblot analyse av RAS proteiner (Figur 4A) viste at uttrykket av KRAS ble økt i KRAS
-amplified mage kreftceller (HSC45, SH101P4 og MKN1), mens verken NRAS eller HRAS ble sterkt uttrykt (Figur 4a, data ikke vist ). Selv om uttrykket av la-7c og la 7g microRNAs har blitt rapportert å regulere RAS uttrykk [8], fant vi lite sammenheng i uttrykket av disse microRNAs med KRAS protein nivåer (Tilleggs fil 12), som foreslo at KRAS overekspresjon i magekreft cellelinjer skyldes først og fremst genomisk forsterkning av KRAS
. Figur 4 Overuttrykte KRAS, og differensial aktivering av KRAS, P44 /42 MAP kinase og AKT i KRAS -amplified magekreftceller. (A) Immunoblot-analyse av ekspresjonsnivåer av KRAS og NRAS i kreftceller. Actin uttrykk ble analysert som en lasting kontroll. (B) Den basale nivået av GTP-KRAS var markert høy i magekreftceller med forsterkede muterte KRAS
(HSC45 og SH101P4). Total lysate (500 ug) ble underkastet en GTP-RAS pull-down-analyse, og GTP-KRAS og GTP-NRAS ble påvist ved immunblot ved anvendelse av anti-KRAS og anti-NRAS antistoffer, henholdsvis. Totale cellelysat (50 ug) ble analysert i parallell for å bestemme nivået av ekspresjon av KRAS og NRAS i celler. (C) GTP-KRAS ble forhøyet etter serum stimulering i MKN1 celler. Celler ble dyrket i vanlig medium inneholdende 10% FCS (N), serum-sultet i 24 timer (-) eller serum-sultet deretter stimulert med 10% FCS i 1 time (+). Totale cellelysat ble underkastet en GTP-KRAS pull-down-analyse. (D) Aktivering av P44 /42 MAP-kinase og AKT i serum-sultet eller stimulert gastrisk kreft celler.
 Perfectus Biomed skal stille ut på IPS -konferansen i Liverpool
Perfectus Biomed skal stille ut på IPS -konferansen i Liverpool
 Anti-coronavirus-molekyler fra mikrober kan være nøkkelen til nye behandlinger
Anti-coronavirus-molekyler fra mikrober kan være nøkkelen til nye behandlinger
 Er C-seksjon bra for barndoms helse?
Er C-seksjon bra for barndoms helse?
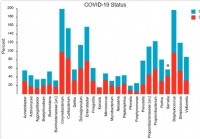 Sammensetning og struktur av nasofaryngeal mikrobiom er relatert til alvorlighetsgraden av COVID-19 sykdom
Sammensetning og struktur av nasofaryngeal mikrobiom er relatert til alvorlighetsgraden av COVID-19 sykdom
 Tarmbakterier knyttet til metabolske endringer og autisme i ny studie
Tarmbakterier knyttet til metabolske endringer og autisme i ny studie
 Plantefôr kan overføre antibiotikaresistente superbugs til mennesker
Plantefôr kan overføre antibiotikaresistente superbugs til mennesker
 Forskning sier i SARS-CoV-2 hundeinfeksjon,
overføring usannsynlig En ny studie publisert på forhåndstrykkserveren bioRxiv* i september 2020 viser at kjæledyrhunder har blitt syke og dør av en mystisk luftveisinfeksjon, som ofte viser tilsted
Forskning sier i SARS-CoV-2 hundeinfeksjon,
overføring usannsynlig En ny studie publisert på forhåndstrykkserveren bioRxiv* i september 2020 viser at kjæledyrhunder har blitt syke og dør av en mystisk luftveisinfeksjon, som ofte viser tilsted
 Ny modell for vaginal mikrobiomtransplantasjon
Bakteriell vaginose er en tilstand som rammer tusenvis av kvinner over hele verden, og er assosiert ikke bare med vaginale symptomer, men med graviditetsrelaterte komplikasjoner inkludert tidlig fødse
Ny modell for vaginal mikrobiomtransplantasjon
Bakteriell vaginose er en tilstand som rammer tusenvis av kvinner over hele verden, og er assosiert ikke bare med vaginale symptomer, men med graviditetsrelaterte komplikasjoner inkludert tidlig fødse
 Forskere utvikler en tilnærming for å vaksinere mot tarmbetennelse
Inflammatorisk tarmsykdom (IBD) er et paraplybegrep som beskriver mange lidelser som involverer kronisk betennelse i fordøyelseskanalen, inkludert ulcerøs kolitt og Crohns sykdom. Disse forholdene er
Forskere utvikler en tilnærming for å vaksinere mot tarmbetennelse
Inflammatorisk tarmsykdom (IBD) er et paraplybegrep som beskriver mange lidelser som involverer kronisk betennelse i fordøyelseskanalen, inkludert ulcerøs kolitt og Crohns sykdom. Disse forholdene er