therapeutische Potenzial von PRL-3-Targeting und klinische Bedeutung der PRL-3
genomischen Amplifikation in Magenkrebs
Zusammenfassung
Hintergrund
Phosphatase Leber-3 der Regeneration (PRL-3) hat die Aufmerksamkeit als eine entscheidende Molekül in den mehreren Stufen der Metastasierung verdient. In der vorliegenden Studie untersuchten wir die Mechanismen PRL-3-Expression regulieren, und bewertet das klinische Potenzial von PRL-3-gezielte Therapie bei Magenkrebs.
Methoden
PRL-3 genomische Amplifikation wurde quantitativ-Polymerase-Ketten analysiert Reaktion und /oder Fluoreszenz-in situ-Hybridisierung in 77 primären Magentumoren. Die Anti-Krebs-Aktivität von PRL-3-Inhibitor (1-4-Brom-2-benzyliden Rhodanin) -Behandlung wurde mit verschiedenen ausgewertet gegen Krebszellen und genetischen Expressionsstatus.
Ergebnisse
PRL-3 genomischen Amplifikation wurde eng konkordant mit hoher Niveau ihrer Proteinexpression in Zelllinien und wurde in 20% (8/40) unter den menschlichen Primärtumoren mit seinen Ausdruck, die waren alle im Stadium III /IV-Krankheit (40%, 8/20), aber in keiner (0 gefunden /37) ohne Ausdruck diejenigen unter. Zusätzlich wurde PRL-3 genomische Amplifikation mit metastasiertem Lymphknoten-Status verbunden sind, im fortgeschrittenen Stadium führt und dadurch schlechte Ergebnisse bei Patienten mit Lymphknotenmetastasen (P = 0,021
). PRL-3 small interfering RNA robust metastatischen Eigenschaften unterdrückt, einschließlich der Zellproliferation, Invasion und Verankerung-unabhängige Koloniebildung. Obwohl weder PRL-3 genomische Amplifizierung noch Expressionsniveau für die Empfindlichkeit auf PRL-3-Inhibitor-Behandlung verantwortlich war, zeigte der Inhibitor dosisabhängige Antikrebswirkung, und bemerkenswert induzierte Apoptose auf allen getesteten Zelllinien mit PRL-3 Expression.
Schlussfolgerungen
Wir waren zum ersten Mal haben gezeigt, dass die PRL-3 genomische Amplifikation eines der vorherrschenden Mechanismen ist seine Expression zu induzieren, vor allem im fortgeschrittenen Stadium, und das PRL-3-gezielte Therapie ein großes Potential gegen Magenkrebs haben mit seinen Ausdruck.
Schlüsselwörter PRL-3 Magenkrebs genomische Amplifikation gezielte Therapie Lymphknoten hintergrund und Magenkrebs (GC) ist die vierthäufigste Krebserkrankung und die zweithäufigste Ursache für Krebs-Todesfälle weltweit [1 ]. Jüngste Verbesserungen in Diagnose-Tools und Verfahren Nachweis von frühen GC erleichtert und dadurch eine ausgezeichnete langfristige Überleben. Doch Patienten mit fortgeschrittener Erkrankung zum Zeitpunkt der Diagnose bleiben schlechte Ergebnisse. Metastasierung ist ein mehrstufiger Prozess, unter Einbeziehung der lokalen Invasion, Verbreitung und Wiederherstellung in entfernte Organe, und ist der wichtigste Faktor für die Mortalität [2]. Daher kann ein besseres Verständnis der Metastasierung öffnen den Weg zu einer Vielzahl innovativer Therapiestrategien in der GC.
Die Protein-Tyrosin-Phosphatasen (PTPs) eine große Familie von Enzymen bilden, die als wichtige regulatorische Komponenten in Signaltransduktionswege dienen [3] . Die Phosphatasen der Leber zu regenerieren (PRL-1, -2 und -3), -Superfamilie zu einer kleinen Klasse von PTP gehören, haben eine einzigartige COOH-terminalen Prenylierung Motiv, das kritisch ihre zelluläre Lokalisation und Funktion beeinflusst [4]. PRL-3 wurde zuerst speziell in Lebermetastasen überexprimiert von Darmkrebs abgeleitet werden identifiziert [5], und anschließend seine Überexpression wurde in verschiedenen Tumortypen dokumentiert, einschließlich GC [6]. PRL-3 kann Krebs Invasion, Migration, das Wachstum und die Angiogenese, entweder durch Dephosphorylierung, die unter der Regie von katalytischen Domäne oder Lokalisierung Plasmamembran katalysiert wird durch COOH-terminalen Prenylierung Motiv [7-9] zu fördern. Somit PRL-3 hat die Aufmerksamkeit als ein entscheidendes Molekül in den mehreren Stufen der Metastasen verdient und damit als neues therapeutisches Ziel. Auf der anderen Seite sind die Mechanismen PRL-3-Expression induzierende nicht vollständig geklärt. Amplifikation genomischer Regionen Onkogene ist der Hauptmechanismus der daraus folgenden Überexpression und der Krebsentwicklung und hat deshalb Bedeutung zielgerichtete Therapien [10] enthält. PRL-3
Genamplifikation Konten teilweise für die Überexpression bei Darmkrebs und Speiseröhrenkrebs [5, 11]. Jedoch bleibt die Beziehung zwischen der genomischen Amplifikation und GC schwer in den beiden mechanistischen und therapeutischen Gesichtspunkten. In der vorliegenden Studie untersuchten wir die Eigenschaften von PRL-3
genomischen Amplifikation in GC und beurteilt ferner das klinische Potenzial von PRL-3-gezielte Therapie.
Methods
Zelllinien und Gewebeproben
die GC-Zelllinie MKN7 wurde aus der Zelle Resource Center for Biomedical Research Institute of Development, Altern und Krebs, Tohoku University (Sendai, Japan) zur Verfügung gestellt. Sieben weitere GC-Zelllinien (GCIY, AZ521, KatoIII, SH10, H111, MKN74 und NUGC4) wurden von RIKEN BioResource Center (Ibaraki, Japan) erworben. Diese Zelllinien decken die zwei Haupttypen von GC [12], Darm-Typ (MKN7, MKN74, AZ521 und H111-Zellen) und diffusen Typ (GCIY, KatoIII, SH10 und NUGC Zellen) [13-15]. MKN7, NUGC4 und AZ521 Zellen wurden von Lymphknotenmetastasen (LNM) gegründet und MKN74-Zellen wurden von Lebermetastasen. KatoIII und GCIY Zellen wurden von metastasierendem Pleuraerguss und Aszites etabliert sind. H111 und SH10 Zellen wurden von der Xenotransplantation hergestellt. Normalen Skelettmuskel C2C12-Zellen wurden von DS Pharma Biomedical Co., Ltd. (Osaka, Japan) erworben. AZ521 und C2C12-Zellen wurden in DMEM-Medium (GIBCO, Carlsbad, CA), supplementiert mit 10% fötalem Rinderserum (FBS) gezüchtet. Die übrigen Zellen wurden in RPMI1640-Medium (GIBCO), ergänzt mit 10% FBS gezüchtet. 1-4-Brom-2-benzyliden Rhodanin wurde von Calbiochem Corp (San Diego, CA) erworben, die als PRL-3-Inhibitors durch einen hohen Durchsatz identifiziert Screening chemischen Bibliothek von Korea Chemical Bank verwenden und hemmte PRL-3-Phosphatase-Aktivität [16]. Tatsächlich Phosphorylierung von KRT8, PRL-3-interacting protein, induziert durch katalytisch inaktive Mutante von PRL-3, aber nicht durch Wildtyp wurde von PRL-3-Inhibitor-Behandlung in einer dosisabhängigen Weise [17] bestätigt. Darüber hinaus Anti-Krebs-Wirksamkeit von PRL-3-Inhibitor-Behandlung zeigte auch in Speiseröhrenkrebs oder Darmkrebs zu der siRNA Behandlung ähnlich zu sein [11, 17].
Von 173 in Formalin fixierten und in Paraffin eingebetteten Gewebeproben Serie, wo wir zuvor PRL-3 Expressionsstatus unter Verwendung immunhistochemischer Färbung (IHC) in GC untersucht [6], 77 Paare von primären Tumorgeweben angepaßt und die entsprechenden normalen Schleimhaut-Gewebe wurden zufällig aus Patienten mit Differenzstufen gemß der 13
th edition ausgewählten der japanischen Klassifikation von Magenkarzinom (JCGC) [18]; 40 Paare mit positiver PRL-3-Expression (10 Patienten im Stadium I, 10 in II, 10 in III und 10 in IV) und 37 Paare mit negativen Ausdruck (10 Patienten im Stadium I, 10 in II, 9 in III, und 8 in IV). Alle Patienten wurden Gastrektomie nach den Magenkrebsbehandlungsrichtlinien in Japan [19] und histopathologische Untersuchungen wurden nach der JCGC getan. Die 6 th Ausgabe der Internationalen Union gegen Krebs (UICC) /TNM-Klassifikation wurde ebenfalls verwendet [20]. Tabelle 1 zeigt die detaillierte Informationen zu 77 Patienten. Alle Gewebeproben wurden an der Kitasato University Hospital gesammelt, und informierte Zustimmung wurde von allen Patienten erhalten. Die vorliegende Studie wurde von der Ethikkommission der Kitasato University.Table 1 Korrelation zwischen PRL-3-Gen-Amplifikation und klinisch-pathologischen Variablen bei 77 Patienten mit Magenkrebs zugelassen
|
PRL-3-Gen-Amplifikation
|
Variablen Bei
Gesamtzahl
Negativität
Positivität
p
Wert
|
|
Anzahl
(%)
Anzahl
(%)
|
PRL-3-Expression
0,006
Negativität
37
37
(100 )
0
(0)
Positivität
40
32
(80)
8 (20)
Alter (Jahre)
0.726
< 60
34
30
(88)
4 (12)
≥60
43
39
(91)
4
(9)
Geschlecht
0.710
männlich
51
45
(88)
6 (12)
Weiblich
26
24
(92)
2 (8)
lymphatischen Permeation
0.343
Abwesenheit
15
15
(100)
0
(0)
Presence
62
54
(87)
8 (13)
Vascular Permeation
0.263
Abwesenheit
25
24
(96)
1 (4)
Presence
52
45
(87)
7
(13)
Differenzierung
0.134
Gut und moderate
31
30
(97)
1 (3)
Schlechte
46
39
(85)
7
(15)
Tiefe der Invasion 0,006 *
T1 (m und sm)
15
15
(100)
0
(0)
T2 (mp und ss)
35
33
(94)
2 (6)
T3 (se)
19
16
(84)
3
(16)
T4 (si)
8 5
(63)
3
(38)
Lymphknotenmetastase
0,022
Abwesenheit
29
29
(100)
0
(0)
Presence
48
40
(83)
8 (17)
JCGC Lymphknotenstatus †
0,004 *
N0
29
29
(100)
0
(0)
N1
21
20
(95)
1 (5)
N2
20
14
(70)
6 (30)
N3 und entfernten Lymphknoten
7
6 (86) seite 1 (14)
UICC Lymphknotenstatus ‡
0.002 *
N0
29
29
(100)
0
(0)
N1
18
17
(94)
1 (6)
N2
16
13
(81)
3
(19)
N3 und entfernten Lymphknoten
14 10
(71)
4 (29)
JCGC stage
0,005 *
I (IA und IB )
20
20
(100)
0
(0)
II 20
20
(100)
0
(0)
III (IIIA und IIIB)
19
15
(79)
4 (21)
IV
18
14
(78)
4 (22)
UICC-Stadium
0,003 *
I (IA und IB)
21
21
(100)
0
(0)
II
20
20
(100)
0
(0)
III (IIIA und IIIB)
16
13
(81)
3
(19)
IV
20
15
(75)
5
(25)
Fluoreszenz in situ Hybridisierungsanalyse
Fluoreszenz in situ Hybridisierung (FISH) Analyse wurde durchgeführt, wie zuvor beschrieben [11]. PRL-3
ist auf Chromosom 8q24.3 (GenBank Zugangsnummer NT 000.008,9) befindet, und das Chromosom 8 Zentromer-Sonde wurde verwendet, die Kopienzahl zu schätzen. Da PRL-3
FISH Scoring-Algorithmen waren nicht standardisiert waren, wurde die Bewertung nach den Kriterien von HER2
basiert [21]. Für jede Probe wurden mindestens 60 Krebszellen erzielt. Positive PRL-3
genomische Amplifikation wurde als Verhältnis der PRL-3
auf Chromosom 8 Zentromer mehr als 2,2 definiert ist, und negativ war das Verhältnis von weniger als 1,8. Wenn das Verhältnis von PRL-3
auf Chromosom 8 Zentromer 1,8 bis 2,2 war, wurden zusätzliche Zellen gezählt, und das Verhältnis von mehr als 2,0 wurde schließlich als positiv [21] betrachtet. Polysomie wurde als die mittlere Chromosom 8 zentromerischen Signale mehr definiert als 3,0 pro Kern [22].
Quantitative-genomischen PCR
Gewebeschnitte von Tumor und dem entsprechenden normalen Schleimhaut, mindestens 5 cm von der Tumorrand erhalten wurden scharf seziert auf Hämatoxylin und Eosin-gefärbten Objektträger, und genomische DNA wurde anschließend unter Verwendung eines QIAamp DNA FFPE Kit (QIAGEN Sciences, Hilden) extrahiert. Quantitative-genomischen Polymerase-Kettenreaktion (Q-PCR) wurde durchgeführt, PRL-3
Genkopie Zahlen mit iQ ™ Supermix (Bio-Rad Laboratories, Hercules, CA) in dreifacher Ausfertigung auf dem iCycler iQ ™ Real-Time PCR-Nachweis zu quantifizieren System (Bio-Rad). Zur Normalisierung der PRL-3
Genkopien pro Zelle, ADAM Metallpeptidase Domäne 2 (ADAM2
, NT 923.907,1), befindet sich auf Chromosom 8p11.2, wurde als endogene Referenz verwendet, weil das Gen-Amplifikation als Kopie definiert ist Anzahl Erhöhung eines begrenzten Bereich eines Chromosoms Arm [10]. ACt Werte wurden als Ct (PRL-3
) berechnet -Ct (ADAM2
) für jede Probe. Relative Kopienzahl wurde bestimmt, wie 2 - ΔΔ Ct, wo ΔΔC t = Δ C t (Tumor) -ΔC t (entsprechend normal) [23]. Die Zunahmen von mehr als 2-fache gegenüber dem entsprechenden normalen wurden als genomische Amplifikation betrachtet. Weitere Datei 1 zeigt detaillierte PCR Zustand und die Sequenzen von Primer und Sonde in der vorliegenden Studie verwendet.
Western-Blotting
Whole Zelllysate wurden in RIPA-Puffer extrahiert (Pierce, Rockford, IL), ergänzt mit 10 &mgr; l /HALT mL ™ Cocktail Kit Protease Inhibitor (Pierce) und HALT ™ Phosphatase-Inhibitor-Cocktail Kit (Pierce), und das Protein wurden auf NuPAGE getrennt ® 4-12% bis-Tris-Gel (Invitrogen) entsprechend dem Protokoll des Herstellers. Sowohl Nachweis und die Quantifizierung der spezifischen Proteine wurden durchgeführt unter Verwendung ATTO Lichtfangs (ATTO Corporation, Tokio, Japan). Zwei kolorektalen Krebszelllinien DLD-1 und SW480-Zellen (RIKEN BioResource) wurden als die niedrige und hohe Expressionskontrollen verwendet wurden, wie zuvor beschrieben [11]
PRL-3 monoklonalen Maus-Antikörper (R &. D Systems, Minneapolis wie beschrieben, MN) und β-Actin-monoklonalen Maus-Antikörper (Sigma, St. Louis, MO) wurden zuvor verwendet [11].
PRL-3 small interfering RNA Transfektion
Zellen mit 1 &mgr; mol /L Accell transfiziert wurden Smartpool, siRNA-PRL-3 (Thermo Fisher Scientific, Lafayette, CO), gemischt mit Accell siRNA Lieferung Medien (Thermo Fisher Scientific) nach dem Thermo Scientific Dharmacons ® Accell ™ siRNA Delivery Protocol [24]. Die Accell Non-Targeting-Pool (siRNA-ctr) und Accell siRNA Lieferung Medien allein wurden als Kontrolle für die nicht-sequenzspezifische Effekte und als Mock-Behandlung verwendet wurden.
Anchorage-unabhängige Koloniebildungstest
verankerungsunabhängiges wurde das Zellwachstum untersucht durch Plattieren 0,36% Top-Agarose (Bacto ™ Agar, Becton, Dickinson and Company, Franklin Lakes, NJ), enthaltend 1 × 10 5 Zellen auf einer Oberfläche von 0,72% Boden Agarose in 6-well Platten [11]. Zellen wurden durch wöchentliche überlagernden frischen Weichagar Lösung zugeführt, und die Kolonien wurden nach 2 Wochen Inkubation photographiert. Die 50% effektive Konzentration (EC 50) -Wert von PRL-3-Inhibitor-Behandlung wurde auf der Grundlage der Messung der Koloniezahl berechnet.
Proliferationsassays und Invasionstest
Der Proliferations-Assay wurde unter Verwendung von Premix-WST 1 Cell Proliferation Assay System (Takara Bio, Tokio). Die Zellen (2 × 10 3) wurden in 96-Well und die Proliferationsaktivität ausgesät wurde durch Absorption bei 450 nm auf ausgewiesenen Beströmungstagen gemessen. Die Empfindlichkeit gegenüber PRL-3-Inhibitor auf antiproliferative bestimmt wurde die 50% ige Hemmkonzentration unter Verwendung von (IC 50) Wert nach der Behandlung für 72 Stunden.
Der Invasionstest wurde in der 24-Well-BD BioCoat ™ Matrigel ™ Invasion durchgeführt Kammer (BD Biosciences Entdeckung Labware, Bedford, MA). Zellen, die durch die Membran wurden in vier getrennten Feldern pro Napf gezählt eingedrungen war. Beide Experimente wurden dreifach durchgeführt.
Apoptosis Assays
Apoptosis-Assays wurden unter Verwendung von Guava PCA-System durchgeführt (Guava Technologies, Inc., Hayward, CA). Zellen (2 × 10 5) wurden 72 Stunden mit der PRL-3-Inhibitors in der angegebenen Konzentration in Medium, ergänzt mit 1,0% FBS behandelt, dann mit Annexin V und 7-AAD (Guava Nexin Reagent) gefärbt. Das Experiment wurde dreifach durchgeführt und analysiert CytoSoft 2.1.5 Software (Guava Technologies).
Statistische Analyse
exakten Fisher-Test oder der Mann-Whitney-U
-Test verwendet wurde, um die Beziehung zwischen statistisch analysieren PRL-3
Gen-Amplifikation und klinisch-pathologischen Variablen. Einweg-Varianzanalyse (ANOVA) mit post-hoc-Test verwendet wurde zwischen drei Gruppen für siRNA-Behandlung (siRNA-PRL-3, siRNA-ctr und spotten) zu vergleichen. Student t
wurde Test zu bewerten therapeutische Wirkung für die einzelnen Konzentrationen von PRL-3-Inhibitor, verglichen mit 0 &mgr; mol /l PRL-3-Inhibitor verwendet. Die Kaplan-Meier-Methode wurde verwendet, kumulative Überlebensrate zu schätzen, und Unterschiede in der Überlebensraten wurden mit der Verwendung des Log-Rank-Test bewertet. Alle Todesfälle von Patienten waren krebsbedingten und krankheitsspezifische Überleben (DSS) wurde ab dem Zeitpunkt der Operation zu dem Zeitpunkt des Todes oder der letzten Follow-up gemessen. P
< 0,05 wurde als statistisch signifikant anzuzeigen. Alle statistischen Analysen wurden mit JMP 7.0-Software (SAS Institute, Cary, NC) durchgeführt.
Ergebnisse | PRL-3-Expression und genomische Amplifikation in Magenkrebs-Zelllinien
Zunächst PRL-3-Expression Status wurde mit bewertet western-Blotting in 8 GC-Zelllinien (1A). PRL-3-Expression in einem nachweisbaren Niveau in allen Zelllinien beobachtet wurde, unter denen fünf Zelllinien (KatoIII, H111, MKN7, MKN74 und NUGC4 Zellen) und 3-Zelllinien (GCIY, AZ521 und SH10-Zellen) zeigte eine hohe und relativ niedrige Expression sind. Anschließend wurde FISH-Analyse zu untersuchen durchgeführt, ob PRL-3-Expression durch die genomische Amplifikation (1B) verursacht wurde. Genomic Verstärkung war offensichtlich positiv in zwei Zelllinien (MKN7 und MKN74-Zellen) und negativ in 6 Zelllinien. 3 der sechs waren dysomic (AZ521, GCIY und NUGC4 Zellen) und drei waren polysomic (KatoIII, SH10 und H111-Zellen). PRL-3
genomische Amplifikation aufgetreten häufig in den verschiedenen Regionen von Chromosom 8, verteilt Einfügungen so genannte, auf Metaphase [10], und war konkordant mit ihrem hohen Ausdruck. Abbildung 1: Expression und genetischen Status von PRL-3 in 8GC Zelllinien. (A) Expression von PRL-3 durch Western-Blot. (B) FISH-Analyse auf Metaphase der PRL-3
Gens (grün). Das Chromosom 8 zentromerischen Sonde (orange) verwendet wurde, die Kopienzahl zu schätzen.
Charakteristisch für PRL-3
genomischen Amplifikation in humanen primären Magenkrebs
In unserer früheren Studie, PRL-3-Expression wurde in 95 festgestellt (55%) von 173 primären GCs von IHC [6]. Um zu untersuchen, wurde die Verbindung zwischen PRL-3-Expression und seiner genomischen Amplifikation, Q-PCR für geführt sowohl den 40 Tumoren mit positiven PRL-3-Expression und 37 Tumoren mit negativem Ausdruck, die von Differenzstufen in den 173 primären Tumoren zufällig ausgewählt wurden. Alle primären Tumoren ohne PRL-3-Expression wurden nicht verstärkt, während 8 (20%) aus den 40 Primärtumoren mit PRL-3-Expression verstärkt wurden (2A). FISH-Analysen bestätigt auch offensichtlich genomische Amplifikation als krebsspezifische Veränderung (2B) und bei fast homogene Muster wiesen sowohl im zentralen Bereich und invasive Bereich innerhalb Tumors. Anschließend wurde die Beziehung zu klinisch-pathologischen Faktoren für PRL-3
genomischen Amplifikation (Tabelle 1) bewertet, wo es wesentlich, nicht nur mit seiner Expression (P
= 0,006), aber auch mit der Tiefe der Tumorinvasion verbunden war ( P
= 0,006), die Anwesenheit von LNM (P
= 0,022), LNM Status (P
= 0,004 in JCGC, P
= 0,002 in UICC) und Stufe (P
= 0,005 in JCGC, P
= 0,003 in UICC). Zusätzlich sind alle primären Tumoren mit genomischer Amplifikation waren im Stadium III oder IV Krankheit (40%, 8/20). Darüber hinaus beeinflusst die genomische Amplifikation negativ auf die Ergebnisse der histologisch node-positive Patienten (P = 0,021
, 2C), obwohl PRL-3-Expression nicht in unserem und anderen früheren Berichten haben [6, 25]. Abbildung 2 Häufigkeit und Prognose von PRL-3 genomischen Amplifikation in 77 menschlichen GC. (A) Häufigkeit der PRL-3
genomischen Amplifikation unter Verwendung von Q-PCR in 77 menschlichen GC. Q-PCR wurde für die beiden 40-Tumoren mit einem positiven PRL-3-Expression und 37 Tumoren mit negativem Ausdruck ausgeführt. PRL-3
Genkopien pro Zelle, ADAM2
, befindet sich auf dem Chromosom 8p11.2, wurde als endogene Referenz verwendet zu normalisieren. ACt Werte wurden als Ct (PRL-3
) berechnet -Ct (ADAM2
) für jede Probe. Relative Kopienzahl wurde bestimmt, wie 2-ΔΔCt, wo ΔΔCt = ACt (Tumor) -ΔCt (entsprechend normal). (B) Repräsentative FISH-Analyse von PRL-3
Gen in Primärtumor und entsprechenden normalen (Fall 82). (C) Kaplan-Meier-Kurven von 5-Jahres-DSS nach der Positivität oder Negativität der PRL-3
genomischen Verstärkung in den histologisch node-positive Patienten. Fehlerbalken, Standardabweichung (SD).
PRL-3 als therapeutisches Ziel konvergenten
GC, die funktionalen Rollen von PRL-3, einschließlich der Invasion und Proliferationsfähigkeiten wurden nur in SGC7901 Zellen dokumentiert worden [25] . Um zu bestätigen, diese metastatischen Eigenschaften unter Verwendung von 3-Zelllinien mit unterschiedlichen PRL-3-Expression und genetischen Status, Knock-down der endogenen PRL-3-Expression wurde unter Verwendung von siRNA-Transfektion durchgeführt; AZ521 Zellen (niedrige Expression und Disomie), H111-Zellen (hohe Expression und Polysomie), MKN74 Zellen (hohe Expression und genomische Amplifikation). Diese Zelllinien wurden mit siRNA-PRL-3 transfiziert oder siRNA-ctr und Western-Blot zeigte die verringerte Höhe der PRL-3-Protein in siRNA-PRL-3-Zellen, aber nicht siRNA-ctr Zellen, verglichen mit Mock-Behandlungszellen ( 3A). Einer der wichtigen Merkmal des metastatischen Phänotyp als die Fähigkeit von Krebszellen soll unter verankerungsunabhängigen Bedingungen [26] zu wachsen, aber die Beteiligung an der PRL-3 bleibt in der GC unbekannt. Alle siRNA-PRL-3-Zellen zeigten die deutlich verringerte Größe und Anzahl der Kolonien, im Vergleich zu siRNA-ctr Zellen oder Mock-Behandlung Zellen (3B). Darüber hinaus im Einklang mit früheren Berichten für andere GC-Zelllinien [25, 27], wir auch bestätigt, dass siRNA-PRL-3-Zellen, die deutlich weniger Proliferationsaktivität (Abbildung 3C) und invasive Fähigkeit (3D) zeigte. Abbildung 3 Hemmung der metastatischen Eigenschaften nach der Transfektion mit siRNA in Zellen mit der Expression. (A) Western-Blot 72 Stunden nach der Transfektion. Der Western-Blot wurde auch quantifiziert. Mock, Behandlung mit Accell siRNA Lieferung Medien allein; ctr, Behandlung mit siRNA-ctr; si, Behandlung mit siRNA-PRL-3. (B) Anchorage-unabhängige Koloniebildung Assays. Repräsentative Bilder der Koloniebildung auf AZ521-Zellen wurden in der oberen Platte gezeigt. Mit Mock-Behandlung als 1,0, ist die relative Rate der Koloniezahl wurde im unteren Bereich angezeigt. Bars, 200 &mgr; m. (C) Proliferationsassay. Die proliferative Aktivität von AZ521 Zellen auf 1, 2, 3, 4 oder 5 Tage nach der Transfektion (oben) und der relativen Proliferationsrate auf 5 Tage nach der Transfektion (unten) gezeigt wurden. (D) Invasionsassay. Die Zellen wurden 4 Tage nach der Transfektion ausgesät, und dann für 22 Stunden inkubiert. Repräsentative Bilder von AZ521 Zellen (oben) und die relative invasive Rate (unten) gezeigt wurden. Bars, 200 &mgr; m; *, P
< 0,05 durch ANOVA mit post-hoc-Test; Fehlerbalken, SD.
therapeutische Potenzial von PRL-3-Inhibitor, 1-4-Brom-2-benzyliden Rhodanin
das therapeutische Potenzial zu bewerten und ein Meilenstein Führung der Reaktion auf PRL-3-gezielte Therapie untersuchen, wir die Anti-Krebs-Aktivität von PRL-3-Inhibitor ausgewertet, zellpermeables Benzyliden Rhodanin-Verbindung [16], gegen 6 Zelllinien mit unterschiedlichen PRL-3-Expression und genetischen Status; GCIY und AZ521 Zellen (niedrige Expression und Disomie), KatoIII Zellen (hohe Expression und Polysomie), SH10-Zellen (niedrige Expression und Polysomie), MKN7 und MKN74 Zellen (hohe Expression und genomische Amplifikation). Zellen wurden mit PRL-3-Inhibitor bei Konzentrationen von 0 bis 50 umol /L im Bereich behandelt. PRL-3-Inhibitor zeigte dosis- und zeitabhängige antiproliferative Wirksamkeit auf allen getesteten Zelllinien, unabhängig von unterschiedlichen PRL-3 Expressionsniveau und genetischen Status und die IC 50 -Werten von GCIY, AZ521, KatoIII, SH10, MKN7 und MKN74-Zellen wurden 26.77, 9.98, 24.26, 23.95, 22.29 und 9.45 &mgr; mol /L, bzw. (4A). AZ521 und MKN74 Zellen waren empfindlicher auf PRL-3-Inhibitor-Behandlung als GCIY und MKN7 Zellen, die als identische Gruppen in Bezug auf die Expression und genetischen Status bzw. kategorisiert wurden. Es wurde nämlich genetische oder Expressionsstatus nicht mit einer Empfindlichkeit von GC-Zellen gegen die PRL-3-Inhibitor in Verbindung gebracht. Ähnliche Wirksamkeit wurde in Verankerung-unabhängige Koloniebildung, und die EG 50 Werte von GCIY, AZ521, SH10 und MKN74 Zellen waren 6,99, 13.05, 9.52, 9.09 mmol /L dargestellt sind (4B). GCIY Zellen empfindlicher Hemmung im Gegensatz zu den anti-Proliferation zeigten. Darüber hinaus auch dieser Inhibitor aufgehoben robust die invasive Fähigkeit von GC-Zellen (4C). Um die Anti-Krebs-Wirksamkeit von PRL-3-Inhibitor-Behandlung zu charakterisieren, wurde die Apoptose-Assay durchgeführt (5A). Obwohl 1 &mgr; mol /l des Inhibitors unzureichend war, die Apoptose über die Basislinie (0 &mgr; mol /L) zu induzieren, 10 umol /L des Inhibitors verursacht robust die drastische Apoptose auf allen getesteten Zelllinien, in denen die 3-fach waren und 11-fach erhöht sich über die Basisregeln in GCIY und MKN74 Zellen. So verdrängte PRL-3-Inhibitor diese metastatischen Eigenschaften auf allen getesteten Zelllinien in dosisabhängig, und weder Expressionsniveau noch genetischen Status zeigte eine klare Korrelation mit der Empfindlichkeit. 4 Anti-Krebs-Aktivität von PRL-3-Inhibitor. (A) Proliferationstest nach der Behandlung mit PRL-3-Inhibitor bei Konzentrationen im Bereich von 0 bis 50 umol /L gegen 6-Zelllinien mit verschiedenen PRL-3 Expression und genetischen Status. Mit mit chemischen Lösung allein behandelten Zellen (0 &mgr; mol /L PRL-3-Inhibitor) als 1,0, ist die relative Proliferationsrate auf 5 Tage nach der Behandlung wurde in der linken Tafel gezeigt. Die proliferative Aktivität von AZ521 Zellen auf 1, 2, 3 oder 4 Tage nach der Behandlung wurde in der rechten Tafel gezeigt. L, niedrige Ausdruck; H, hoher Expression; D, Disomie; P, Polysomie; A, Verstärkung; IC50, die 50% ige inhibitorische Konzentration. (B) Anchorage-unabhängige Koloniebildung Assays. Repräsentative Bilder der Koloniebildung auf AZ521 Zellen (oben) und die relative Geschwindigkeit der Koloniezahl (unten) gezeigt wurden. Bars, 200 &mgr; m; EC50, die 50% ige wirksame Konzentration. (C) Invasionsassay. Repräsentative Bilder (oben) und die relative invasive Rate (unten) gezeigt wurden. Bars, 200 &mgr; m; *, P
< 0,05 von Student t
Test, verglichen mit 0 &mgr; mol /l PRL-3-Inhibitor; Fehlerbalken, SD.
Abbildung 5 PRL-3-Inhibitor-vermittelte Apoptose. (A) Apoptosis Assay wurde mit PRL-3-Inhibitor (0 bis 10 umol /L) nach der Behandlung 72 Stunden durchgeführt. Repräsentative Zahlen der Apoptose-Assay auf AZ521 und C2C12-Zellen wurden in der linken Tafel, und der Prozentsatz und SD der frühen Apoptose (unten rechts Quadrant) und späte Apoptose (Quadrant oben rechts) sind in jeder Platte dargestellt. Mit mit chemischen Lösung allein behandelten Zellen (0 &mgr; mol /L PRL-3-Inhibitor) 1,0, die relativ spät Apoptoserate nach der Behandlung wurde in der rechten Tafel gezeigt. *, P
< 0,05 von t
Testschüler, verglichen mit 0 &mgr; mol /l PRL-3-Inhibitor. (B) PRL-3-Inhibitor-Behandlung gegen normale Skelettmuskel C2C12-Zellen. C2C12 Zellen niedriger Expression von PRL-3 zeigte als GC-Zellen mittels Western Blot. Proliferationstest nach der Behandlung mit PRL-3-Inhibitor durchgeführt wurde. Die Fehlerbalken, SD.
Schließlich untersuchten wir, ob PRL-3-Inhibitor Zytotoxizität in normalen Skelettmuskulatur induziert, wo PRL-3 vorwiegend exprimiert wird [28]. Beide Proliferation und Apoptose-Assays wurden unter Verwendung von normalem Skelettmuskel C2C12 Zellen, die mit dem Inhibitor behandelt durchgeführt und zeigte, dass 10 &mgr; mol /L des Inhibitors antiproliferative und apoptotische Antwort auf C2C12 im Gegensatz zu den Wirksamkeiten auf allen getesteten GC-Zelllinien zu veranlassen, ist fehlgeschlagen ( 5A und 5B).
Diskussion
wie LNM als wichtiger prognostischer Faktor für GC betrachtet wird [29], der Forschung der verursachenden Moleküle reflektieren LNM ist ein vielversprechender Weg, die Ergebnisse zu verbessern. Die enge Verknüpfung von LNM mit PRL-3-Expression hat daher Potenzial als neues therapeutisches Ziel [6, 25]. Allerdings haben sich die Kriterien für die PRL-3-gezielte Therapie nicht nachgewiesen worden ist, und es ist wichtig, die Eigenschaften von PRL-3
genomischen Verstärkung in den beiden mechanistischen und therapeutischen Gesichtspunkten, weil der Hauptmechanismus der zu klären konsequente Ausdruck und die Entwicklung von Krebs [10]. In der vorliegenden Studie bieten wir die wichtige Hinweise für die Entwicklung dieser Therapiestrategie gegen GC.
Die Beziehung zwischen PRL-3-Expression und seine genomische Amplifikation waren noch nie so weit untersucht worden. PRL-3
genomische Amplifikation mit seiner Expression Status in Zelllinien concordant war, und wurde in 20% (8/40) unter primären humanen Tumoren mit Expression, die waren alle im Stadium III oder IV Krankheit (40%, 8 /gefunden 20), aber in keiner (0/37) unter denen, ohne Ausdruck. Zusätzlich PRL-3
genomische Amplifikation wurde mit LNM Status verbunden sind, im fortgeschrittenen Stadium führt und dadurch schlechte Ergebnisse bei Patienten mit LNM (P
= 0,021). So PRL-3
genomische Amplifikation kann die relevante Veränderung für LNM sein, und eine der vorherrschenden Mechanismen seinen Ausdruck in der fortgeschrittenen Stadium zu induzieren. Allerdings sind die meisten Tumoren exprimieren PRL-3 wurden nicht amplifiziert, insbesondere im earler Stufe. In embryonalen Maus-Fibroblasten-Zellen, die mit Wildtyp, aber nicht p53 - /-, PRL-3 ist in einer p53-abhängigen Weise induziert [30]. Die p53-Mutation oder Verlust der Funktion, jedoch hat in allen GC-Zelllinien in der vorliegenden Studie verwendet dokumentiert worden, mit Ausnahme von NUGC4 Zellen (Die TP53 Web-Site http:.. //P53 frei fr /) anzeigt, daß ein anderer Mechanismus dort unabhängig von p53-Weg ist. PRL-3-Expression wurde berichtet durch mitogene Cytokine wie IL-6, IL-21, HGF oder IGF-1 in Myelomzelllinien [24] oder als TGF-β in Kolon-Krebszelllinien auf Transkriptionsebene reguliert wird [ ,,,0],31]. Vor kurzem PolyC-RNA-binding protein 1 (PCBP1) wurde als Translationsregulator der PRL-3 [32] identifiziert. Die alternativen Mechanismen auf Transkriptions oder Translationsebene kann PRL-3-Expression beteiligt sein zu regulieren.
Wir bestätigten auch, dass siRNA-vermittelte PRL-3 Knockdown signifikant die Zellproliferation und die Invasion im Einklang mit früheren Berichten für andere GC-Zelllinien unterdrückt [25 27], und darüber hinaus zum ersten Mal offenbart die verringerte Wirkung der Koloniebildung unter verankerungsunabhängigen Bedingungen unterstützen, dass PRL-3 attraktive therapeutische Ziel gegen GC sein kann.
|  Strategische Vorbereitung der pädiatrischen Gesundheitsversorgung auf die zweite Welle der COVID-19-Pandemie
Strategische Vorbereitung der pädiatrischen Gesundheitsversorgung auf die zweite Welle der COVID-19-Pandemie
 Führt Bluthochdruck immer zu schwerem COVID-19?
Führt Bluthochdruck immer zu schwerem COVID-19?
 Antioxidantien in der Nahrung können das Darmkrebsrisiko erhöhen.
Antioxidantien in der Nahrung können das Darmkrebsrisiko erhöhen.
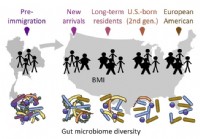 Die Migration beeinflusst die Darmmikrobiota, was sich wiederum auf die Gesundheit auswirkt, finden Forscher
Die Migration beeinflusst die Darmmikrobiota, was sich wiederum auf die Gesundheit auswirkt, finden Forscher
 Ist der Kaiserschnitt gut für die Gesundheit von Kindern?
Ist der Kaiserschnitt gut für die Gesundheit von Kindern?
 Das Lungenmikrobiom sagt den Schweregrad der COVID-19-Krankheit voraus
Das Lungenmikrobiom sagt den Schweregrad der COVID-19-Krankheit voraus
 GSK-3-Inhibitoren sind vielversprechend bei der Behandlung von Coronavirus-Infektionen
Forscher in den Vereinigten Staaten haben einen neuen Ansatz zur Behandlung von Infektionen mit Coronaviren wie dem schweren akuten respiratorischen Syndrom Coronavirus 2 (SARS-CoV-2) vorgeschlagen –
GSK-3-Inhibitoren sind vielversprechend bei der Behandlung von Coronavirus-Infektionen
Forscher in den Vereinigten Staaten haben einen neuen Ansatz zur Behandlung von Infektionen mit Coronaviren wie dem schweren akuten respiratorischen Syndrom Coronavirus 2 (SARS-CoV-2) vorgeschlagen –
 Bestimmte Bakterienarten können das HIV-Risiko bei Frauen erhöhen,
findet neue Studie Eine kürzlich in der veröffentlichte Studie Die Lancet-Infektionskrankheiten, beschreibt sieben Arten von Vaginalbakterien, die das Risiko einer HIV-Infektion bei Frauen signifik
Bestimmte Bakterienarten können das HIV-Risiko bei Frauen erhöhen,
findet neue Studie Eine kürzlich in der veröffentlichte Studie Die Lancet-Infektionskrankheiten, beschreibt sieben Arten von Vaginalbakterien, die das Risiko einer HIV-Infektion bei Frauen signifik
 Alkohol schädigt das Mikrobiom im Mund
Eine neue Studie hat gezeigt, dass Alkohol das natürliche Bakterienmilieu im Mund verändert und schädigt. Die Studie mit dem Titel „Das Trinken von Alkohol wird in einer großen Studie an amerikanische
Alkohol schädigt das Mikrobiom im Mund
Eine neue Studie hat gezeigt, dass Alkohol das natürliche Bakterienmilieu im Mund verändert und schädigt. Die Studie mit dem Titel „Das Trinken von Alkohol wird in einer großen Studie an amerikanische