Estratto
16S ribosomiale recenti gene RNA (rRNA) profiling molecolare della mucosa dello stomaco ha rivelato una complessità sorprendente microbiota. Helicobacter pylori Visto:. Li XX, Wong GL-H, per KF, Wong VW-S, Lai LH, Chow DK-L, et al. (2009) batterica Microbiota Profiling nella gastrite senza Helicobacter pylori Editor: Niyaz Ahmed, Università di Hyderabad, India Received: 4 giugno 2009; Accettato: 27 ottobre, 2009; Pubblicato: 24 nov 2009 Copyright: © 2009 Li et al. Questo è un articolo ad accesso libero distribuito sotto i termini della Creative Commons Attribution License, che permette l'uso senza restrizioni, la distribuzione e la riproduzione con qualsiasi mezzo, a condizione che l'autore originale e la fonte sono accreditati Finanziamento:. Questo lavoro è supportato da l'Università cinese di Hong Kong. I finanziatori avevano alcun ruolo nel disegno dello studio, la raccolta e l'analisi dei dati, la decisione di pubblicare, o preparazione del manoscritto Competere interessi:.. Gli autori hanno dichiarato che non esistono interessi in competizione Introduzione Commensal microbiota è parte integrante di un essere umano [1]. La stragrande maggioranza dei microbi abitano il nostro tratto gastrointestinale, con più di 800 specie provenienti da nove batterica e una phyla archaeal. Questo variegato microbiota contribuisce a sventrare la maturazione [2], [3], [4], la nutrizione ospite e resistenza ai patogeni [5]. I microbi anche interagire direttamente con ospite umano regolando la proliferazione epiteliale intestinale, il deposito di grasso e le risposte infiammatorie [3], [6], [7]. Mentre alcuni microbi possono da sola causare gravi malattie, molte condizioni croniche sono probabilmente dovute a perturbazioni del microbiota generale. Ad esempio, le allergie e l'asma sono legati all'infanzia uso di antibiotici, che possono alterare microbiota intestinale [8]. Altre condizioni associate con microbiota intestinale includono l'autismo ad insorgenza tardiva [9], malattia infiammatoria intestinale [10], e il cancro [11]. Tradizionalmente, i metodi di coltivazione-based sono utilizzati per ottenere ceppi microbici per un'ulteriore caratterizzazione. Tali studi hanno fornito il fondamento della nostra comprensione della microbiologia. Tuttavia, la coltivazione è spesso alta intensità di lavoro e può fallire per molti microbi. osservazione microscopica è anche usato per stimare l'abbondanza di microbi, e in misura limitata, assegna microbi a taxon [12]. Recentemente, 16S gene RNA ribosomiale (rRNA) i profili di sequenza vengono utilizzati per spiegare la diversità microbica, spesso al livello phylotype. Utilizzando 16S rRNA sequencing, i microbi dalla bocca [13], [14], esofago [15], stomaco [16], intestino tenue [17], del colon [18], [19] e la vagina [20] sono state studiate. Questi studi hanno esplorato la diversità microbica all'interno del corpo umano e ha rivelato una vasta popolazione di microbi incolti e non caratterizzate, che era stato sfuggente per i metodi di coltivazione basati. Attraverso questi high-throughput 16S rRNA sequencing e altri sforzi di sequenziamento metagenomiche, perturbazioni microbiota sono stati trovati ad essere associata con la malattia parodontale [21] e l'obesità [22], [23], [24]. Nello stomaco , acidità gastrica uccide molti microbi ingerito. E 'stato generalmente considerato che lo stomaco non è abitabile da qualsiasi microbo fino alla scoperta di Helicobacter pylori In questo studio, siamo andati a indagare ulteriormente i potenziali associazioni tra i cambiamenti microbiota di stomaco e non H. pylori Risultati albero Taxon analisi Abbiamo analizzato il corpo e dell'antro biopsie da 5 individui normali e 5 NHNN individui antrale gastrite (tutte le femmine, di pari età) . Tutti i pazienti erano H. pylori Per indagare la rappresentazione complessiva dei diversi livelli taxon nel biota stomaco, abbiamo costruito un albero taxon (Figura 1 e Figura supplementare S1). È interessante notare che, ogni phylum è stato dominato solo da uno o due livelli inferiori (taxon classe, ordine, famiglia o genere). È un dato di fatto, ogni phylum stato dominato solo 1-2 generi. Ad esempio, i Firmicutes phylum più abbondanti è rappresentato da 383 cloni, di quei 333 cloni sono dalla classe Bacilli. Successivamente, 273 cloni erano dall'ordine Lactobacillales. Duecentocinquanta quattro cloni erano dalla famiglia Streptococcaceae. E tutti i 254 cloni sono stati dal genere Streptococcus la ricchezza e la diversità Quando è stato utilizzato l'intero set di dati (1223 cloni), la copertura di Good era 96%, indicando che quattro filotipi supplementari ci si aspetterebbe per ogni 100 cloni supplementari da sequenziare. Questo livello di copertura indicato che la maggior parte delle sequenze batteriche erano presenti nei cloni sequenziati. La diversità di stima da stime versione 8.0 ha indicato che circa 200 filotipi possono essere presenti nei campioni di biopsia allo stomaco umano (supplementare figura S2). Abbiamo stimato ulteriormente la ricchezza di specie in quattro differenti campioni di biopsia (NMA (Normal Antrum), NMB (Corpo normale), AGA (Antral gastrite Antrum), AGB (Antral gastrite corpo); supplementare Tabella S2). Ricchezza di specie non era differente tra le biopsie antrali gastrite e biopsie normali (p > 0.1, spaiato t-test) confronto microbiota batterica tra due diverse sedi anatomiche (antro e corpo) in pazienti normali Firmicutes phylum e Streptococcus sulla base del 1223 sequenze 16S rRNA, il phylum Firmicutes è stato il phylum più abbondante con 383 cloni. Il phylum Proteobacteria era un secondo vicino con 345 cloni. È interessante notare che il phylum Firmicutes era più abbondante nelle biopsie antrali gastrite che in biopsie normali (41% vs. 22%, tabella 1), mentre il phylum Proteobacteria era più abbondante nelle biopsie normali (37% vs. 20%). Dal 16S rRNA sequencing è il costo proibitivo per un campione più ampio, abbiamo sviluppato un approccio quantitativo in tempo reale specifiche taxon PCR (qPCR) per quantificare l'abbondanza di Firmicutes e Streptococcus diciassette coppie aggiuntive di campioni antro e la biopsia del corpo di pazienti normali e 18 coppie aggiuntive di campioni antro e la biopsia del corpo di pazienti gastrite antrale sono stati analizzati mediante Firmicutes-specifiche qPCR. Totalmente, 90 biopsie (46 campioni di pazienti gastrite 23 antrali e 44 campioni provenienti da 22 pazienti normali) sono stati analizzati (tabella 2). L'età media dei pazienti gastrite antrale era 67,6 ± 11,4 (mediana: 69, range: 46-86), mentre l'età media dei controlli è stato 58,3 ± 14,7 (mediana: 52, range: 40-87). L'età media del gruppo normale era più giovane di quella del gruppo gastrite antrale. Ma l'analisi statistica ha dimostrato che non vi era alcuna correlazione tra età e Firmicutes o Streptococcus Per gli stessi campioni bioptici, il genere Streptococcus il semplice rilevamento di sequenze di geni 16S rRNA non implica che batteri vivi sono presenti o batteri sono infatti residenti invece di passanti nello stomaco. Abbiamo quindi condotto due esperimenti supplementari. In primo luogo, abbiamo tentato la coltivazione di batteri dalle biopsie. Una condizione di coltura adatto per Streptococcus in secondo luogo, 14 campioni bioptici sia da gastrite antrale e normali pazienti sono stati lavati in tampone fosfato (PBS) con tre condizioni sempre più difficili. Se le condizioni di lavaggio dure non rimuovono i batteri dalla biopsia, è suggestivo che questi batteri attaccano strettamente alla mucosa dello stomaco. Oltre il 90% dei batteri totali rimasta attaccata alle biopsie dopo tre lavaggi consecutivi con condizioni sempre più dure (supplementare Figura S5A). L'ultima fase di lavaggio è stato fatto ad alta potenza su una macchina desktop vortice. Allo stesso modo, la maggior parte del Streptococcus In questo studio, abbiamo profilato il microbiota batterica nelle biopsie gastriche appaiati (antro e corpo) da normali e antrali pazienti gastrite. Tutti i pazienti sono H. pylori Dato che rigorose fasi di lavaggio non erano in grado di separare il microbiota dalle biopsie, ipotizziamo che la maggioranza dei batteri identificati sono associati strettamente con la mucosa dello stomaco. Inoltre, siamo stati in grado di coltivare maggior parte del Streptococcus Per apprezzare ulteriormente la complessità generale del microbiota stomaco, abbiamo costruito un albero taxon sulla base dei cloni identificati di guardare in ogni livello taxon tra cui phylum, classe, ordine, famiglia e genere. È interessante notare che, abbiamo scoperto che per ogni phylum, uno o due generi sono stati prevalentemente presenti. I generi cinque più comuni tra cui Streptococcus È interessante notare che la profilazione 16S rRNA ha rivelato una significativa sovrarappresentazione del phylum Firmicutes (soprattutto a causa della sovra-rappresentazione del Streptococcus Sia l'aumento di Streptococcus gastriche campioni di biopsia Questo studio è stato approvato dal l'Università cinese di Hong Kong comitato etico di ricerca clinica. Tutti i pazienti hanno firmato un consenso informato per ottenere i campioni di studio. Due biopsie della mucosa gastrica (antro e corpo dello stomaco) sono stati raccolti da ciascun paziente durante l'endoscopia di routine presso il Prince of Wales Hospital di Hong Kong. Per evitare la contaminazione, una nuova pinza endoscopia sterilizzati è stato utilizzato quando si scatta una seconda biopsia dello stesso paziente. Le biopsie sono state scatto congelato in ghiaccio secco e conservati a -80 ° C. I pazienti che assumono antibiotici o FANS (definiti come qualsiasi uso di FANS per almeno una settimana negli ultimi 3 mesi precedenti l'endoscopia) o sono risultati positivi per H. pylori Costruzione di librerie di cloni 16S rRNA e sequenziamento Totale DNA genomico è stato isolato dalle biopsie utilizzando il kit di DNA mini (Qiagen, Valencia, CA, USA ) con vetro battere metodo battitore come precedentemente descritto [16]. Due controlli negativi con solo acqua sterile sono stati estratti utilizzando lo stesso protocollo. Le concentrazioni di DNA estratti sono stati misurati con NanoDrop 1000 Spettrofotometro (Thermo Scientific, Minneapolis, MN, Stati Uniti d'America). Due universali 16S batteriche rRNA primer, B8F20 [35] (5'-AGAGTTTGATCCTGGCTCAG-3 ') e B806R20 [36] (5'-GGACTACCAGGGTATCTAAT-3') sono stati utilizzati per amplificare la regione che corrisponde alla posizione 8-806 del Escherichia coli L'analisi filogenetica e la diversità microbica stima Il test chimerico utilizzando il server Bellerofonte (http.: //foo.maths.uq.edu.au/~huber/bellerophon.pl) [37] è stato utilizzato per testare potenziali sequenze chimerici. Un clone è stato trovato per essere chimerico e successivamente esclusa. Poi, le sequenze 1223 nonchimeric sono stati analizzati mediante RDP II (ribosomiale Database Progetto II) classificatore (http://rdp.cme.msu.edu/classifier/classifier.jsp) sulla base di un ingenuo Bayesiano rRNA classificatore [38]. Base strumento di ricerca locale di allineamento (BLAST) fornito da geni verdi (http://greengenes.lbl.gov/cgi-bin/nph-blast_interface.cgi) è stata effettuata per trovare le sequenze più simili nel database. Abbiamo usato il 97% di identità di sequenza come cutoff per filotipi definiscono [39]. Sequenze con identità < 97% per le sequenze esistenti nel database sono stati considerati romanzo. L'albero taxon è stato costruito utilizzando il risultato classificazione del classificatore RPD II. stimatore Chao1 del 8 programma stima (http://viceroy.eeb.uconn.edu/estimates) è stato utilizzato per stimare la diversità microbica. Il metodo di Buono è stato utilizzato per calcolare la copertura di sequenziamento [40]. Real-time PCR quantitativa (qPCR) primer e sonde Q-PCR sono stati progettati sulla base delle sequenze ottenute dalle librerie clonati. In primo luogo abbiamo allineato tutte le sequenze clonate di ClustalW (http://www.ebi.ac.uk/Tools/clustalw2/index.html) con i parametri di default. I primer qPCR erano B8F20 e B801R21 (5'-ACCAGGGTATCTAATCCTGTT-3 '). Le sequenze di sonda MGB sono: Sonda generici (VIC) 5'- CAGCAGCCGCGGTAA-3 ', la sonda Firmicutes (FAM) 5'- AAGATTCCCTACTGCTGCCT-3' e Streptococcus in teoria, l'abbondanza di un taxon è 2 -ddCt. Streptococcus Abbiamo ottenuto 32 biopsie supplementari da 16 pazienti per coltura batterica. Le biopsie sono stati messi in tampone fosfato (PBS, pH = 7,2) e tagliato in piccoli pezzi con un bisturi. I campioni sono stati poi spalmate su piastre di agar sangue (CM331, Oxoid, Basingstoke, UK) con sangue di cavallo al 5%. Le piastre sono state poste in un CO 5% 2 incubatore a 37 ° C per 24 ore. Le colonie con emolisi su agar sangue sono state raccolte per 16S rRNA sequenziamento. Per la prova biopsia lavaggio, sono stati raccolti 14 campioni supplementari da entrambi i pazienti gastrite antrali e persone normali. Ogni campione è stato posto in una provetta da 2,0 ml e lavato 3 volte (200 microlitri di PBS per ogni lavaggio) in condizioni sempre più difficili. Il primo lavaggio è stato fatto da un leggero scuotimento mano. Il surnatante è stato trasferito in uscita. Nuovo PBS è stato aggiunto al biopsie per ulteriore lavaggio. Per il secondo e il terzo lavaggio, i tubi sono stati in agitazione dal miscelatore tubo trio TM-2F (All-lab scientifica, AU) di grado 3 e grado 6 livello di potenza, rispettivamente, corrispondente all'incirca vortex dolce e vigorosa. Poi duplex real-time PCR è stata effettuata per testare i batteri totali e il Streptococcus test del chi-quadro di Pearson è stato utilizzato per confrontare i numeri di clone di diversi taxa tra i diversi gruppi di campioni (NMA: antro normale, NMB: corpo normale, AGA: antrale gastrite antro, AGB: corpo gastrite antrale) dal risultato di sequenziamento 16S rRNA, quando il numero clone per ogni gruppo campione era di almeno 10. test di ANOVA è stato utilizzato per confrontare i dati di abbondanza batterica da qPCR saggi. Tutte le analisi sono state effettuate utilizzando SPSS per Windows, versione 11.5 (SPSS Inc., Chicago, IL, USA). P < 0,05 è stato considerato statisticamente significativo. Per confrontare la ricchezza di specie tra normale e antrali pazienti gastrite, il numero phylotype effettivo per ogni paziente è stato contato prima. Spaiato t-test è stato poi utilizzato per confrontare i due gruppi di pazienti.
infezione e non-steroidei anti-infiammatori uso (FANS) sono due collaboratori principali di gastrite e ulcera peptica. Tuttavia, poco si sa circa l'associazione tra gli altri membri del microbiota stomaco e le malattie gastriche. In questo studio, la clonazione e il sequenziamento del 16S rRNA è stato utilizzato al profilo del microbiota stomaco di pazienti normali e gastrite. Sono stati identificati centotrenta tre filotipi da otto phyla batterica. Il microbiota stomaco è stato trovato essere strettamente aderito alla mucosa. Undici Streptococcus
filotipi sono stati coltivati con successo dalle biopsie. Da uno a due generi rappresentati la maggioranza dei cloni in una delle phyla identificato. Abbiamo inoltre sviluppato due test PCR quantitativa in tempo reale per quantificare l'abbondanza relativa del phylum Firmicutes e Streptococcus
genere. Significativamente più alta abbondanza del phylum Firmicutes e Streptococcus
genere all'interno del phylum Firmicutes è stata osservata nei pazienti con gastrite antrale, rispetto ai controlli normali. Questo studio suggerisce che il livello di genere taxon può in gran parte rappresentano taxa molto più alti, come il phylum. La rilevanza clinica e il meccanismo alla base della composizione del microbiota alterata nella gastrite richiedono ulteriori studi funzionali
infezione o non-steroidei anti-infiammatori uso di droghe. PLoS ONE 4 (11): e7985. doi: 10.1371 /journal.pone.0007985
e la sua associazione con la gastrite e ulcera peptica [25]. Altro che pochi altri Helicobacter
specie [26], [27], [28], non si aspettava che lo stomaco conterrebbe molti altri microbi vivi. acidità ridotto a causa di gastrite atrofica progressiva può aumentare la diversità microbica [29]. Uno studio condotto da Monstein et al.
Utilizzando temporale gradiente di temperatura elettroforesi su gel (TTGE) e una piccola scala 16S rRNA sequencing suggerito altri microbi come Enterococcus
, Pseudomonas
, Streptococcus
, Staphylococcus
e Stomatococcus
erano presenti nello stomaco [30] anche. Un recente su larga scala 16S rRNA sforzo più sequenziamento identificato 128 filotipi da 8 phyla in 23 pazienti americani del Nord [16]. È interessante notare la presenza di H. pylori
nello stomaco non ha influenzato la composizione complessiva del microbiota a livello di phylum.
e non FANS gastrite (NHNN). Abbiamo ipotizzato che quando H. pylori
non è presente, altri gruppi batterici /specie possono contribuire o essere associati con lo sviluppo gastrite. A livello microbiota, anche noi vorremmo risolvere un problema chiave nel campo: a quale taxon profondità (s) fa il microbiota appaiono relativamente stabili in modo tale che le perturbazioni a questi livelli possono essere rilevanti per la salute umana? Abbiamo usato 16S rRNA gene clonazione e il sequenziamento al profilo del microbiota stomaco dal normale e NHNN gastrite pazienti, e real-time PCR quantitativa (qPCR) test specifici taxon di quantificare l'abbondanza relativa del phylum Firmicutes e Streptococcus
genere.
negativi con entrambi i test di all'ureasi rapida e 16S rRNA sequencing. Nessuno dei pazienti aveva assunto FANS entro 6 mesi prima di subire l'endoscopia. Almeno 60 cloni di ogni biopsia (corpo o antro, quindi almeno 120 cloni di ogni individuo) sono stati sequenziati usando un'ampia gamma 16S rRNA PCR prodotti. Un totale di 1223 non H. pylori
sequenze microbiche sono stati ottenuti. Questi microbi appartengono a 8 phyla (133 filotipi), di cui sono presenti in una vasta maggioranza 5 phyla (Firmicutes, Bacteroidetes, Actinobacteia, Fusobatteri e Proteobacteria) (1211 out del 1223, o 99,0%). Sono stati identificati nove filotipi con similarità di sequenza inferiore al 97% per le sequenze presenti nelle banche dati pubbliche. Sei di questi 9 filotipi erano rappresentati da singoli cloni (Tabella supplementare S1).
. I generi cinque più comuni tra cui Streptococcus
(254 cloni), Prevotella
(243), Neisseria
(175), Haemophilus
(122) , Porphyromonas
(68), ha costituito il 70,5% di tutti i cloni microbiche.
<. p> Nei pazienti normali, è stata osservata alcuna differenza significativa tra i due sedi anatomiche (antro e corpo) per uno dei gruppi taxon, fatta eccezione per la famiglia Prevotellaceae e il genere Prevotella
dove i valori di p (chi di Pearson Test -square) erano tra 0,01-0,05 (Figura 1).
genere sono stati arricchiti nello stomaco di antrali gastrite pazienti
(Figura 2). I dati qPCR specifici taxon per Firmicutes erano altamente correlati con i dati di sequenziamento 16S rRNA per i suddetti 20 campioni bioptici (2 biopsie per ognuno dei 5 normali e 5 pazienti antrale gastrite) (Figura supplementare S3).
abbondanza (p > 0,1) (Figura supplementare S4). Questi campioni sono stati divisi in 4 gruppi, antrale gastrite antro (AGA), antrale corpo gastrite (AGB), antro normale (NMA), e per il corpo normale (NMB). L'abbondanza di Firmicutes era significativamente più alta in AGA che in NMA o NMB (One-way ANOVA (analisi della varianza) di prova, p = 0.004 ep = 0.046, rispettivamente) e in AGB che in NMA (solo andata ANOVA, p = 0.039 ) (Figura 3A). è stata osservata alcuna differenza significativa tra AGA e AGB (solo andata ANOVA, p = 0,855), o NMA e NMB (p = 0.832).
è stato analizzato anche da Streptococcus
SPECIFICI qPCR. Streptococcus
abbondanza era 72% e il 76% più elevata nei AGA contro rispettivamente NMA o NMB, e il 66% e il 70% più elevata nei AGB contro NMA o NMB, rispettivamente (Figura 3B). I valori di p per test di ANOVA sono mostrati nella Figura 3B. Simile al test Firmicutes, è stata osservata alcuna differenza significativa tra AGA e AGB (solo andata ANOVA, p = 0,999), o NMA e NMB (p = 0,999).
Streptococcus
coltivazione e la biopsia lavaggio
è stato utilizzato dal momento che sembravano essere sovrarappresentati nei pazienti gastrite antrali. Sedici coppie (antro e corpo) di biopsie sono stati utilizzati per la cultura sulle piastre di agar sangue. Colonie sono stati sequenziati per il gene 16S rRNA per l'identificazione. Undici filotipi di Streptococcus
sono stati isolati (supplementare S3 tabella). Questi 11 filotipi costituivano il 93,3% (o 237 di 254 cloni) di tutti i cloni individuati nella vasta gamma 16S rRNA sequencing, che indica che la maggior parte del Streptococcus
filotipi sono vivi nelle biopsie gastriche.
batteri rimasti attaccati alle biopsie dopo le 3 fasi di lavaggio (Figura supplementare S5B).
Discussione
negativo e senza uso di FANS. Attraverso un'ampia gamma 16S rRNA sequenziamento del gene, abbiamo identificato 1.223 non H pylori
batteri cloni, simile ad un precedente studio (studio Bik, 1.056 non H pylori
batteri cloni) [16 ]. Anche se i due studi hanno analizzato due geograficamente (Hong Kong vs California) ed etnico (cinese vs caucasici, ispanici e afro-americana) popolazioni divergenti, le complessità generale microbiota sono sorprendentemente simili (Tabella 3). Entrambi gli studi hanno identificato circa 130 (133 e 127 per questo e lo studio Bik, rispettivamente) filotipi da sette a otto phyla. La maggior parte dei cloni (77,4% di questo studio e il 79,8% di studio Bik) sono state condivise. I due più abbondante generi ( Streptococcus
e Prevotella
) erano identiche. Questi due generi hanno rappresentato il 40,6% e il 41,5% di tutti i cloni in questo studio e lo studio Bik. Entrambi gli studi hanno indicato che circa 200 differenti filotipi possono essere presenti nella mucosa dello stomaco. Tale somiglianza drammatica tra i due studi mette in evidenza la pressione selettiva per il microbiota in ambiente stomaco duro. Inoltre, abbiamo trovato poca differenza in microbiota batterica tra i due siti anatomici (antro e corpo) in pazienti normali, nonostante la rilevanza clinica per il campionamento bioptico in diverse sedi anatomiche [31].
filotipi identificati mediante un'ampia gamma 16S rRNA sequencing, suggerendo che questi batteri può essere vera residenti nella mucosa dello stomaco.
(phylum Firmicutes), Prevotella
e Porphyromonas
(Bacteroidetes), così come Neisseria
e Haemophilus
(Proteobacteria), costituito il 70,5% di tutti i cloni microbiche.
genere all'interno del phylum) e una sottorappresentazione del phylum Proteobacteria nelle biopsie di pazienti gastrite antrale. Abbiamo sviluppato un approccio qPCR specifico per taxon per analizzare l'abbondanza del Firmicutes e streptococco
taxa per 90 biopsie (46 campioni provenienti da 23 pazienti gastrite antrali e 44 campioni provenienti da 22 pazienti normali) e ha confermato l'eccessiva presenza di questi due taxa in antrale gastrite stomaco del 42% e 71%, rispettivamente. La maggior parte del Streptococcus
filotipi individuate dal sequenziamento erano batteri alfa-emolitico che sono potenziali patogeni (ad esempio, Streptococcus pneumoniae
, Streptococcus mitis
e Streptococcus salivarius
). Alcuni Streptococcus
specie sono resistenti alle condizioni di pH bassi e possono sopravvivere nello stomaco [32]. Il nostro esperimento dati di coltivazione e di lavaggio anche suggerito che questi erano effettivamente vivere, biota residente nello stomaco.
abbondanza è causale per la gastrite antrale o di una conseguenza del cambiamento ambientale locale a causa di gastrite antrale è rimasta a cui rispondere. Un approccio potenziale sta usando modello murino privo di germi [33]. Un'altra domanda interessante è che se certe composizioni microbiota proteggere, o, in alternativa, sensibilizzare la mucosa dello stomaco da agenti patogeni invasori come H. pylori
. Infine, le nuove tecnologie di sequenziamento ad alto rendimento sono suscettibili di fornire dati più completi sul microbiota da diverse sedi anatomiche lungo il tratto digestivo umano e in diversi momenti [34].
Materiali e metodi
da test rapido dell'ureasi (RUT) o il test istologico sono stati esclusi. la demografia del paziente è mostrato nella Tabella 2.
16S rRNA gene. I 25 miscele microlitri PCR incluse 1 × tampone PCR compreso 1,5 mm MgCl 2 (Qiagen), 20 mM cloruro di tetrametilammonio, 0.1 mm di ciascun dNTP, 0,4 mM di ciascun primer, 1 unità di polimerasi HotStar Taq DNA (Qiagen), e 2 ml estratto il DNA. A trentacinque ciclo di PCR è stata effettuata per amplificare il frammento di 799 bp. I prodotti di PCR sono stati controllati mediante elettroforesi su gel di agarosio. Per ogni prodotto, una singola banda potrebbe essere osservata ai raggi UV, mentre nessun gruppo è stato visto per i controlli negativi. I prodotti 16S rRNA sono stati purificati con Sephadex G-50 colonne (Sigma-Aldrich, St. Louis, MO, USA), legatura con vettori T e trasformato in E. coli
cellule JM109 utilizzando il semplice sistema vettore pGEM-T (Promega, Madison, WI, USA). Abbiamo selezionato 5 pazienti (10 campioni di biopsia) con gastrite antrale e 5 controlli normali (10 campioni di biopsia) per la costruzione di 20 16S librerie gene rRNA. Per ciascuna libreria biopsia gastrica, almeno 60 colonie sono stati selezionati per il sequenziamento. I prodotti di PCR sono stati sequenziati usando BigDye terminator v3.1 kit ciclo di sequenziamento (Applied Biosystems, Foster City, CA, USA). Le reazioni di sequenziamento utilizzando B8F20 come primer di sequenziamento sono state effettuate su un sequenziatore ABI 3730xl (Applied Biosystems)
sonda (FAM) 5'- TACACATGGAATTCCAC-3 ' . Per misurare l'abbondanza di un taxon specifico, due sonde (una specifica per il taxon di interesse e la seconda generici per tutti i batteri) sono stati utilizzati nello stesso amplicone PCR. (Figura 2). La miscela di 25 ml di PCR incluso 1 × Buffer A, 3,5 mm MgCl 2, 200 micron dNTP con dUTP al posto di dTTP, 400 Nm di ciascun primer, 100 nM di ogni sonda, 0,01 U /mL Uracil-N-Glycosylase, e 0,05 U /mL TaqGold (Applied Biosystems). Per il saggio Firmicutes-specifico, la condizione bicicletta era: 1) 50 ° C per 2 min; 2) 95 ° C per 10 min; 3) 40 cicli di 95 ° C per 20 sec, 58 ° C per 15 sec, 70 ° C per 80 sec. Per il Streptococcus
saggio SPECIFICI, la condizione di ciclismo era: 1) 50 ° C per 2 min; 2) 95 ° C per 10 min; 3) 40 cicli di 95 ° C per 20 sec, 57 ° C per 1 min, 70 ° C per 1 min. Il Streptococcus
16S rRNA frammento (DQ346438) clonato nel facile vettore pGEM-T è stato utilizzato come standard per la qPCR (sia per i Firmicutes e Streptococcus
saggi) nel 7500 ABI reale -time sistema PCR (Applied Biosystems). A causa di qualche lieve differenza tra le sonde generici e specifici taxon, ciclo delta soglia delta (DDCT) è stato utilizzato per indicare l'abbondanza del taxon specifica in tutta la popolazione di batteri (Ct TSU:. Ct della sonda specifica taxon da campioni non noti, Ct BUU: Ct della sonda universale batterico da campioni sconosciuti, Ct TSS: Ct del taxon sonda specifica dallo standard plasmide, Ct BSS: Ct della sonda universale batterica dallo standard plasmide)
coltivazione
biopsia lavaggio
quantità nei surnatanti PBS delle tre fasi di lavaggio e le biopsie lavati.
Statistiche
Informazioni di supporto
Figura S1. Albero genealogico taxon dettagliata
doi: 10.1371 /journal.pone.0007985.s001
(2.00 MB TIF)
Figura S2.
Specie stima ricchezza
doi: 10.1371 /journal.pone.0007985.s002
(0.97 MB TIF)
Figura S3.
Correlazione tra qPCR e 16S rRNA clonazione e il sequenziamento
doi: 10.1371 /journal.pone.0007985.s003
(0.98 MB TIF)
Figura S4.
mancanza di correlazione tra l'età del paziente e Firmicutes o Streptococcus abbondanza
doi: 10.1371 /journal.pone.0007985.s004
(2.01 MB TIF)
Figura S5.
lavaggio Harsh non rimuove i batteri dalle biopsie
Doi: 10.1371 /journal.pone.0007985.s005
(1.90 MB TIF)
Tabella S1.
Novel phyltoypes
doi: 10.1371 /journal.pone.0007985.s006
(0.03 MB XLS)
Tabella S2.
Specie stima ricchezza in diversi campioni di biopsia
Doi: 10.1371 /journal.pone.0007985.s007
(0.02 MB XLS)
Tabella S3.
doi: 10.1371 /journal.pone.0007985.s008
(XLS 0.02 MB)
 Umani contro virus:possiamo evitare l'estinzione nel prossimo futuro?
Umani contro virus:possiamo evitare l'estinzione nel prossimo futuro?
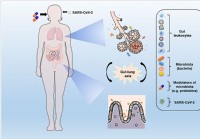 La modulazione del microbiota e il ripristino dell'eubiosi potrebbero aiutare a frenare le complicanze del COVID-19
La modulazione del microbiota e il ripristino dell'eubiosi potrebbero aiutare a frenare le complicanze del COVID-19
 Farmaco per la perdita di peso Wegovy approvato dalla FDA
Farmaco per la perdita di peso Wegovy approvato dalla FDA
 La qualità del sonno potrebbe essere un indicatore per un futuro studio sulla malattia di Alzheimer
La qualità del sonno potrebbe essere un indicatore per un futuro studio sulla malattia di Alzheimer
 Buone notizie per chi soffre di IBS poiché i ricercatori identificano il "prurito intestinale"
Buone notizie per chi soffre di IBS poiché i ricercatori identificano il "prurito intestinale"
 Il Simposio Scientifico di LABVOLUTION si concentra su questioni chiave nelle scienze della vita
Il Simposio Scientifico di LABVOLUTION si concentra su questioni chiave nelle scienze della vita
 La disbiosi nel microbiota intestinale può causare una grave infezione secondaria nei pazienti COVID-19
Un interessante studio condotto da scienziati negli Stati Uniti ha recentemente rivelato che la comunità microbica nellintestino è direttamente interessata dalla sindrome respiratoria acuta grave coro
La disbiosi nel microbiota intestinale può causare una grave infezione secondaria nei pazienti COVID-19
Un interessante studio condotto da scienziati negli Stati Uniti ha recentemente rivelato che la comunità microbica nellintestino è direttamente interessata dalla sindrome respiratoria acuta grave coro
 La ricerca mostra che le infestazioni da parassiti intestinali riducono la gravità del COVID-19
Stiamo imparando di più sulla malattia COVID-19 ogni giorno. Gli adulti di qualsiasi età con determinate condizioni mediche di base sono a maggior rischio di malattie gravi dovute al virus che causa i
La ricerca mostra che le infestazioni da parassiti intestinali riducono la gravità del COVID-19
Stiamo imparando di più sulla malattia COVID-19 ogni giorno. Gli adulti di qualsiasi età con determinate condizioni mediche di base sono a maggior rischio di malattie gravi dovute al virus che causa i
 Nuovo modello per il trapianto di microbioma vaginale
La vaginosi batterica è una condizione che colpisce migliaia di donne in tutto il mondo, ed è associato non solo a sintomi vaginali, ma anche a complicanze legate alla gravidanza, tra cui travaglio pr
Nuovo modello per il trapianto di microbioma vaginale
La vaginosi batterica è una condizione che colpisce migliaia di donne in tutto il mondo, ed è associato non solo a sintomi vaginali, ma anche a complicanze legate alla gravidanza, tra cui travaglio pr