: Familial cancer gastrique et Chordome dans la même famille
cancers gastriques de Résumé sont le deuxième cancer le plus fréquent dans le monde et représentent un fardeau important pour toutes les sociétés même si l'incidence de la maladie est en baisse dans le monde industrialisé. L'étiologie de la maladie est complexe et semble être principalement due à des facteurs environnementaux, mais une faible proportion de cas sont reconnus comme étant associés à des facteurs génétiques. Deux formes héréditaires de cancer de l'estomac ont été identifiées, celle qui est associée à clusterings familiaux de cancer de l'estomac et l'autre étant un sous-groupe de familles qui appartiennent à la non polyposis colorectal cancer héréditaire (ou syndrome de Lynch). Dans ce rapport, nous présentons une petite famille nucléaire qui est inhabituel en ce qu'il est un regroupement de malignité qui comprend le cancer de l'estomac, le cancer colorectal et chordome. L'analyse génétique n'a révélé aucune mutation causale dans les gènes associés à HNPCC ou E-cadhérine. Ensemble, le tableau clinique de cette famille peut indiquer que d'autres facteurs génétiques sont derrière le regroupement de cette famille de malignité.
Mots-clés
cancer de l'estomac HNPCC E-cadhérine chordome Présentation
cancers gastriques représentent le deuxième cancer le plus fréquent dans le monde entier, bien que l'incidence de la maladie semble diminuer dans le monde industrialisé. cancers gastriques sont morphologiquement hétérogène avec trois types; diffuser le cancer gastrique, un glandulaire (ou intestinale) type et un mélange diffus et la maladie intestinale.
Les causes du cancer de l'estomac sont à la fois l'environnement et la génétique ou un mélange des deux. Relativement récemment une cause génétique définitive du cancer gastrique (mutations dans le gène E-cadhérine) a été identifié, d'abord dans une grande Nouvelle-Zélande Maori Kindred [1] et par la suite dans d'autres familles gastriques [2]. Les patients présentant des mutations dans le gène de la E-cadhérine ont tendance à présenter une maladie diffuse alors qu'aucun des facteurs génétiques ont été impliqués dans des agrégats familiaux de cancer gastrique intestinal.
Une cause génétique alternatif de cancer de l'estomac, en association avec une variété d'autres cancers épithéliaux est cancer colorectal non polypose héréditaire (HNPCC). Cette entité est caractérisée par un cancer colorectal précoce et une variété d'autres cancers épithéliaux, y compris cancer de l'endomètre et le cancer de l'estomac. La base génétique de cette condition est une ventilation de mismatch repair due à des mutations dans les gènes qui contrôlent ce processus. Actuellement, quatre gènes ont été isolés et associés à HNPCC, hMLH1, hMSH2, et hMSH6 hPMS2. Les deux hMSH2 et hMSH6 résident sur le chromosome 2, hMLH1 est sur le chromosome 3 et hPMS2 est sur le chromosome 7 (pour revue voir Niessen et al 2004 [3]).
Chordomas sont rares lents tumeurs osseuses en croissance qui sont censées découler de restes notochorde [3]. Ils ont tendance à se produire à la base du crâne et ont un aspect histologique relativement bénin. En dépit de leurs chordomes apparence bénignes ont des propriétés infiltrants qui sont difficiles à contrôler. Il y a une légère prépondérance pour les hommes d'être affectés par un mâle rapport global femelle d'environ 1,7: 1. La prépondérance des hommes devient plus frappant chez les patients avec chordomes sacral où le ratio homme-femme se rapproche de 3: 1. On sait peu sur la base moléculaire de chordomes, car il semble y avoir aucune anomalie chromosomique grave ou tout autre élément distinctif distinct. Deux loci génétiques ont été identifiés. La première indication d'une base génétique pour chordome provenaient d'études de liaison qui ont révélé un locus du gène sur le chromosome 1 [4]. Un second locus a été identifié dans une petite série de familles liée au chromosome 7q33, qui a révélé un certain nombre de gènes candidats potentiels [5].
Dans ce rapport, nous avons identifié une petite famille qui se caractérise par le cancer de l'estomac et de la présence . de chordome dans l'un des trois frères et sœurs qui ont tous succombé à la malignité
patients et méthodes
l'histoire familiale de la maladie est la suivante: le proband présenté à l'âge de 56 ans avec un adénocarcinome peu différencié du caecum sans la présence de polypes adénomateux. Au moment de l'excision de la tumeur a métastasé aux trois ganglions lymphatiques. Le patient a été constaté que la maladie métastatique généralisée à l'âge de 60 ans et est mort un an plus tard. La seule autre découverte remarquable par rapport à ce patient était la aplasie congénitale de la cuisse gauche. L'histoire de la famille de l'proband a révélé une histoire de cancer gastro-intestinal (voir Fig. 1). Figure 1 Arbre généalogique de la famille. cancer St = estomac; CH = chordome; CRC = cancer colorectal. Squares représentent les mâles, les femelles cercles
Le père de la proband a été diagnostiqué avec le cancer de l'estomac à l'âge de 67 ans. La tumeur a été diagnostiquée comme un carcinome mucineux indifférencié avec des cellules annulaires chevalière qui avaient au moment de la chirurgie déjà propagé au foie.
Le frère aîné du patient a été diagnostiqué à l'âge de 7 ans 1/2 avec un chordome maligne à la base de son crâne et mourut peu après. La tumeur était située entre le clivus et pons qui a donné lieu à des signes neurologiques typiques. La tumeur avait empiété sur le nerf optique entraînant une atrophie sévère, en particulier sur le côté droit. En outre, il y avait de multiples métastases hépatiques.
Le second frère a été diagnostiqué avec un adénocarcinome de l'estomac d'origine dans le cardia. La tumeur était un type mucineux indifférenciée entrecoupé de cellules en bague chevalière, similaire à celle du père. Au moment du diagnostic de la maladie a été trouvée être devenue généralisée avec des métastases aux vertèbres, le foie, les glandes surrénales et les poumons.
Analyse CLHPD
Les séquences codantes entières de hMSH2, hMLH1 et E-cadhérine, y compris l'intron /exon les limites ont été criblées pour des mutations par analyse CLHPD. Toutes les conformations inhabituelles ont ensuite été évaluées par un séquençage direct de l'ADN.
Réaction en chaîne par polymérase (PCR) pour l'analyse de CLHPD a été réalisée en utilisant des amorces spécifiques de hMSH2 et hMLH1 comme décrit précédemment (Holinski-Feder et al, 2001). La réaction consistait en 1,0 uM de chaque amorce, 1 U de Taq Platinum (Gibco-BRL), 2-5 mM de MgCl
2 et 200 μ
M de chaque dNTP.
L'amplification par PCR a été réalisée par une dénaturation initiale à 94 ° C pendant 5 minutes, suivi par 14 cycles de 94 ° C pendant 1 min, 7 ° C plage de toucher des roues pendant 1,5 min et 72 ° C pendant 2 min, puis 20 cycles en utilisant une température de recuit de 0,5 ° C inférieure à la partie inférieure du plage touché. L'étape de recuit a été réalisé comme un protocole de touché avec une gamme de 7 ° C, diminuant de 0,5 ° C /cycle de plus de 14 cycles. Ceci a été suivi par une étape d'extension finale à 72 ° C pendant 10 min, une étape de dénaturation finale à 95 ° C pendant 5 min et une étape de recuit lent de 95 ° C à 65 ° C pendant 30 min pour favoriser la formation d'heteroduplex. La PCR a été réalisée sur un express PCR (Hybaid) instrument équipé d'un couvercle chauffant pour éviter l'utilisation de l'huile minérale.
Analyse CLHPD a été réalisée en utilisant un système Varian Helix (Varian Inc., Walnut Creek, CA). Les produits de PCR (2-5 μ
l) ont été injectées directement dans un Eclipse ADN (Hewlett Packard) ou colonne d'hélice (Varian) et on élue de la colonne en utilisant un gradient croissant d'acétonitrile et à une température du four de la colonne appropriée pour chaque exon de hMSH2 , hMLH1 et E-cadhérine (températures réelles disponibles sur demande). Heteroduplex formés au cours de la PCR d'un échantillon hétérozygote ont été détectées en tant que pic d'élution supplémentaire avant le pic homoduplex. La détection d'heteroduplex a été rendue plus simple avec l'utilisation de logiciels d'examen de CLHPD fourni par Varian. Les températures de fusion prévue des doubles produits d'ADN brin ont été obtenus en utilisant le programme CLHPD-MELT disponible à partir de http:... //Www insertion stanford edu /fondre html (pour plus de détails, voir les tableaux 1a et 1b. ).
Pour chaque segment un fragment de contrôle négatif (amplifié à partir d'ADN isolé à partir d'un donneur sain et normal qui n'a pas d'antécédents familiaux de maladie) a été exécuté à travers la colonne de dénaturation à la température non dénaturant de 50 ° C. 50 ° C profil de pic a ensuite été comparé au profil de température de fusion du fragment de Stanford respectif, ainsi que trois incréments de 1 ° C de part et d'autre de la température de fusion prédite. Partiellement conditions dénaturées ont été établies lors d'un déplacement dans le temps de rétention d'au moins ou égal à 30 secondes à travers une gamme C d'incrément de 1 ° a été faite. La température de fusion optimale a toujours été considérée comme la plus haute température, dans des conditions de dénaturation partielle qui ne présentent pas de dégradation de profil.
Séquençage d'ADN
Tous les hétéroduplexes ont été soumis à un séquençage d'ADN pour déterminer la variation génétique précise sur une unité de mise en séquence semi-automatique ( modèle 310, Perkin-Elmer Applied Biosystems Division, Foster City, CA) en utilisant didésoxy. Le séquençage des produits de PCR a été réalisée en utilisant la version 1 BigDye séquençage didésoxy kit Ready Rxn (Perkin-Elmer, Foster City, CA):. Résultats
La famille représentée sur la figure. 1 est conforme aux critères d'Amsterdam II où d'autres tumeurs malignes épithéliales peuvent remplacer le cancer colorectal. La présence de deux cancers gastriques est inhabituelle et pourrait être évocateur d'un cancer gastrique familial due à des mutations dans la E-cadhérine.
Il n'y avait pas de matériel disponible pour les tests immunohistochimie ou l'instabilité des microsatellites. Un échantillon d'ADN est disponible à partir du propositus qui a été soumis à une mutation d'analyse. Depuis la famille a adhéré aux critères d'Amsterdam II initialement hMSH2 et hMLH1 analyse de mutation a été entreprise. Aucune mutation ont été identifiés dans la séquence codante soit hMSH2 ou hMLH1 y compris les introns /limites d'exons.
Comme il y avait deux personnes atteintes de cancer de l'estomac d'un écran plus de mutation a été entreprise dans le gène E-cadhérine, mais aucun changement délétères ont été identifiés de. Rapport
l'incapacité à identifier des mutations dans hMLH1, hMSH2 ou E-cadhérine suggère qu'il existe probablement d'autres facteurs génétiques sous-jacents de la maladie dans cette famille. Il n'y a toutefois la possibilité qu'une mutation a été manquée dans l'un de ces trois gènes puisque l'analyse de suppression n'a pas été effectuée, ni qu'il a été possible de le faire car il y avait un matériel génétique insuffisante pour permettre que ceci se produise. En outre, étant donné que les patients ont succombé à leurs maladies, il y a plusieurs décennies, il est peu probable que les tests immunohistochimie ou l'instabilité des microsatellites pourrait être effectuée pour évaluer la probabilité de cette famille appartenant à l'entité de HNPCC. E-cadhérine était un candidat probable en raison de la présence de cellules en bague chevalière dans les deux cancers de l'estomac.
Il reste plusieurs possibilités en ce qui concerne ce qui peut avoir eu lieu au sein de cette petite famille. Tout d'abord, il reste possible que ce soit en effet une famille de HNPCC non seulement parce qu'il peut y avoir des mutations dans les deux hMLH1 ou hMSH2 mais aussi nous n'avons pas examiner hMSH6 ou hPMS2. Le gène hPMS2 reste un candidat probable qu'il a été associé à des cas héréditaires récessives du syndrome de Turcot [9]. En outre, hPMS2 réside sur le chromosome 7 et est entouré par des pseudogènes, dont certains sont exprimés quoique à des niveaux nettement inférieurs à ceux de type sauvage hPMS2. Depuis un nouveau locus pour chordome a également été identifié sur le chromosome 7, il reste la possibilité que, dans cette famille ces deux résultats ne sont pas sans rapport. Malheureusement, nous ne saurons jamais la réponse à cela car il y a du matériel restant insuffisant pour étudier à partir du propositus et les échantillons de tumeur prélevés sur les autres membres de la famille ne sont plus disponibles. En ce qui concerne le père du proband nous ne savons pas si l'aplasie congénitale de la cuisse gauche a été liée à la maladie dans cette famille.
En résumé, cette famille ne représente pas seulement un défi par rapport à l'identification d'une prédisposition génétique mais aussi, si les membres de la famille étaient vivants aujourd'hui, pour le conseil génétique.
 Transmission mère-enfant du SRAS-CoV-2 pendant la grossesse possible mais rare,
Transmission mère-enfant du SRAS-CoV-2 pendant la grossesse possible mais rare,
 Si vous avez plus de 50 ans,
Si vous avez plus de 50 ans,
 La fonction hépatique peut être importante dans le risque de maladie d'Alzheimer
La fonction hépatique peut être importante dans le risque de maladie d'Alzheimer
 La supplémentation en acides gras à chaîne courte améliore la récupération de l'AVC,
La supplémentation en acides gras à chaîne courte améliore la récupération de l'AVC,
 Les probiotiques comme thérapie adjuvante pour les patients COVID-19
Les probiotiques comme thérapie adjuvante pour les patients COVID-19
 Selon une étude, les produits de nettoyage peuvent augmenter le risque d'asthme chez les enfants
Selon une étude, les produits de nettoyage peuvent augmenter le risque d'asthme chez les enfants
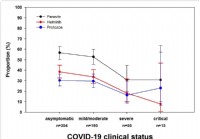 La recherche montre que les infestations de parasites intestinaux réduisent la gravité du COVID-19
Nous en apprenons chaque jour davantage sur la maladie COVID-19. Les adultes de tout âge atteints de certaines conditions médicales sous-jacentes courent un risque accru de maladie grave due au virus
La recherche montre que les infestations de parasites intestinaux réduisent la gravité du COVID-19
Nous en apprenons chaque jour davantage sur la maladie COVID-19. Les adultes de tout âge atteints de certaines conditions médicales sous-jacentes courent un risque accru de maladie grave due au virus
 Certaines espèces bactériennes peuvent augmenter le risque de VIH chez les femmes,
trouve une nouvelle étude Une étude récente publiée dans le The Lancet Maladies Infectieuses, décrit sept espèces de bactéries vaginales qui peuvent augmenter considérablement le risque dinfection
Certaines espèces bactériennes peuvent augmenter le risque de VIH chez les femmes,
trouve une nouvelle étude Une étude récente publiée dans le The Lancet Maladies Infectieuses, décrit sept espèces de bactéries vaginales qui peuvent augmenter considérablement le risque dinfection
 Le sang jeune redonne de la vitalité aux personnes âgées
Le Dracula de Bram Stoker a survécu grâce au sang de jeunes filles. Maintenant, les chercheurs ont découvert quil y avait peut-être une part de vérité dans cette théorie bizarre ! Selon une génétici
Le sang jeune redonne de la vitalité aux personnes âgées
Le Dracula de Bram Stoker a survécu grâce au sang de jeunes filles. Maintenant, les chercheurs ont découvert quil y avait peut-être une part de vérité dans cette théorie bizarre ! Selon une génétici