 Une personne atteinte d'un phéochromocytome présente généralement trois symptômes classiques :maux de tête, transpiration et palpitations cardiaques (un rythme cardiaque rapide) en association avec une tension artérielle nettement élevée (hypertension).
Une personne atteinte d'un phéochromocytome présente généralement trois symptômes classiques :maux de tête, transpiration et palpitations cardiaques (un rythme cardiaque rapide) en association avec une tension artérielle nettement élevée (hypertension).
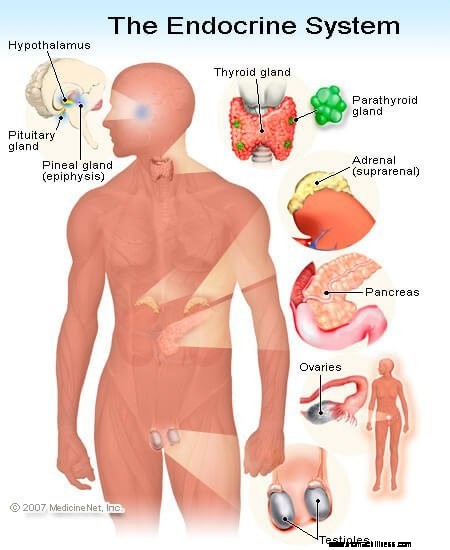
Les phéochromocytomes sont un type de tumeur des glandes surrénales qui peuvent libérer des niveaux élevés d'épinéphrine et de noradrénaline. Comme son nom l'indique, les glandes surrénales sont situées près de la zone "rénale" (rein). Une glande surrénale se trouve au-dessus de chacun des deux reins.
Malgré leur petite taille, les glandes surrénales ont de nombreuses fonctions. Ce sont des glandes endocrines complexes (sécrétant des hormones). Les cellules de différentes régions des glandes surrénales ont des fonctions différentes dans le système endocrinien. La partie externe de la glande surrénale s'appelle le cortex surrénalien. Dans une partie du cortex surrénal (zona fasciculata et zona reticularis), les cellules sécrètent du cortisol, une hormone similaire à la cortisone, qui est essentielle pour gérer le stress. Dans une autre zone (zona glomerulosa), les cellules sécrètent une hormone appelée aldostérone qui aide à la régulation de l'eau et du sel. et le contrôle de la pression artérielle.
La zone interne de la glande surrénale est appelée médullosurrénale. C'est là que les cellules sécrètent des substances appelées catécholamines - épinéphrine, noradrénaline et dopamine. Ce sont des hormones « de fuite ou de combat ». Ils sont en partie responsables de cette sensation de montée d'« adrénaline » que les gens ressentent lorsqu'ils ont peur. Ce sont ces cellules qui sont surproduites par un phéochromocytome. Fondamentalement, un phéochromocytome est une tumeur de ces cellules sécrétant des catécholamines, et qui provoque les signes et symptômes cliniques dont nous parlerons ci-dessous. Les cellules sécrétant des catécholamines sont parfois appelées cellules chromaffines et se trouvent dans d'autres régions du corps ainsi que dans la médullosurrénale.
Parfois, les phéochromocytomes proviennent de cellules chromaffines situées à l'extérieur de la glande surrénale. Dans ce cas, ils sont appelés phéochromocytomes extra-surrénaliens ou paragangliomes et sont généralement situés dans l'abdomen.
Les phéochromocytomes peuvent survenir chez les personnes de tout âge. L'incidence maximale se situe entre la troisième et la cinquième décennie de la vie. Les phéochromocytomes sont, heureusement, assez rares, et la plupart d'entre eux sont bénins.
L'hypertension artérielle, ou hypertension, survient le plus souvent sans aucun symptôme et a pour cette raison été qualifiée de "tueur silencieux". L'hypertension artérielle non compliquée peut persister pendant des années, voire des décennies, sans provoquer de symptômes. Cependant, lorsque des complications de la maladie commencent à se développer en raison de dommages au système vasculaire, des symptômes peuvent survenir.
En savoir plus sur les causes de l'hypertension artérielle »
Une personne atteinte d'un phéochromocytome présente généralement trois symptômes classiques :maux de tête, transpiration et palpitations cardiaques (rythme cardiaque rapide) associés à une pression artérielle nettement élevée (hypertension). Les autres affections pouvant accompagner ces symptômes classiques sont les suivantes :anxiété, nausées, tremblements, faiblesse, douleurs abdominales et perte de poids.
Certaines personnes, cependant, ne développent jamais les symptômes d'un phéochromocytome. Jusqu'à 10 % des cas sont découverts fortuitement, ce qui signifie qu'ils ne sont pas suspectés et ne sont découverts que lorsque le patient subit des études diagnostiques pour d'autres affections. Dans certains cas, l'hypertension artérielle va et vient et peut être difficile à documenter. Dans d'autres cas, la pression artérielle est constamment élevée et facilement enregistrée.
Les phéochromocytomes ne sont présents que chez environ 0,2 % de toutes les personnes souffrant d'hypertension artérielle. Il existe certaines conditions, cependant, dans lesquelles le diagnostic de phéochromocytome peut figurer en bonne place sur la liste des possibilités; ils sont discutés ci-dessous.
Les phéochromocytomes peuvent être une composante de certains syndromes familiaux ou génétiques. La maladie familiale la plus courante est appelée néoplasie endocrinienne multiple, ou NEM en abrégé. Deux types de MEN - MEN 2A et 2B - sont associés aux phéochromocytomes. Les deux sont des syndromes génétiques familiaux et se transmettent de parent à enfant de manière autosomique dominante.
Les phéochromocytomes ne sont pas les seules tumeurs qui surviennent dans les NEM 2A et 2B. MEN 2A comporte un risque accru de tumeurs des parathyroïdes, des glandes proches de la thyroïde qui aident à réguler les niveaux de calcium dans le corps. Et les NEM 2A et 2B augmentent le risque de carcinome médullaire de la thyroïde - un type inhabituel de cancer de la thyroïde. Dans les familles où le MEN est suspecté, des tests génétiques peuvent être effectués pour aider à identifier les membres de la famille à risque.
Les phéochromocytomes sont une caractéristique d'autres troubles génétiques, notamment le syndrome de von Hippel-Lindau et la neurofibromatose de type 1. Ces deux troubles sont associés au développement de nombreuses tumeurs bénignes et malignes.
Il existe également de nombreuses personnes atteintes de phéochromocytomes sans antécédents familiaux connus. Ces cas sont qualifiés de sporadiques. En général, si ces patients ont une maladie bilatérale (phéochromocytomes dans les deux glandes surrénales) ou sont diagnostiqués avant l'âge de 21 ans, un dépistage génétique est recommandé.
Fondamentalement, tout ce qui peut provoquer une suractivité du système nerveux sympathique peut figurer sur la liste des diagnostics à exclure lors de la suspicion d'un phéochromocytome. Le système sympathique est le panneau de contrôle principal régissant la libération de la réponse «fuite ou combat» en réponse au stress ou à la peur, comme mentionné ci-dessus. Les choses qui peuvent stimuler cela incluent les médicaments (même l'utilisation excessive de décongestionnants doit être envisagée); le sevrage des médicaments (comme l'arrêt soudain de certains médicaments contre l'hypertension ); les attaques de panique et les lésions de la moelle épinière font partie des nombreuses conditions qui peuvent également entraîner certains des symptômes observés dans les phéochromocytomes.
Le phéochromocytome est une possibilité chez toute personne présentant la triade classique de symptômes - maux de tête, transpiration et palpitations cardiaques - en particulier en cas d'hypertension artérielle (bien que l'hypertension artérielle ne soit pas toujours présente). Un médecin devient plus méfiant si le patient est jeune et n'a pas d'autres facteurs de risque ou d'habitudes pouvant causer ces résultats.
Peut-être que le patient connaît bien ses antécédents familiaux et peut informer son médecin d'autres types de tumeurs endocrines, y compris les phéochromocytomes diagnostiqués chez des membres de la famille. Par conséquent, un médecin soupçonnant un phéochromocytome familial peut passer directement à des tests génétiques. Cependant, dans la plupart des cas, si la suspicion est élevée, le médecin procède à une série de tests pour mesurer les hormones coupables et leurs produits de dégradation (métabolites) énumérés ci-dessous.
Premièrement, les hormones telles que les catécholamines et les métanéphrines sont mesurées dans une collecte d'urine de 24 heures, et les métanéphrines peuvent également être mesurées dans le sang. Si ceux-ci sont supérieurs à 2 fois le niveau normal, des examens d'imagerie sont généralement effectués pour examiner les glandes surrénales.
Si l'imagerie par IRM ou tomodensitométrie des glandes surrénales montre une masse dans la glande (ou à l'extérieur), une intervention chirurgicale peut être effectuée. S'il n'est pas clair que la masse est réellement fonctionnelle et liée aux résultats cliniques, ou s'il n'y a pas de masse visible à l'imagerie, un autre test peut être effectué. Ce test appelé balayage 131-I-MIBG est assez spécifique pour les phéochromocytomes. Dans ce test, une molécule d'iode radioactif est injectée dans la circulation sanguine et localisée dans la zone de la tumeur, permettant la visualisation du phéochromocytome sur des études d'imagerie.
La chirurgie est le traitement définitif. Jusqu'à ce que la tumeur soit retirée, le contrôle de la pression artérielle est une priorité absolue. Le contrôle de la pression artérielle avant et pendant la chirurgie est la partie la plus délicate des soins. Il existe un risque de développer une crise hypertensive aiguë (une augmentation potentiellement dangereuse, soudaine et sévère de la pression artérielle) après l'administration d'une anesthésie pendant la chirurgie. La pression artérielle est donc gérée avec des médicaments spéciaux avant et pendant la chirurgie et est soigneusement surveillée tout au long de la procédure. Une consultation avec un endocrinologue est recommandée pour aider à concevoir un traitement pour chaque patient.
En raison des types d'hormones impliquées dans le phéochromocytome, les premières tentatives de contrôle de la pression artérielle utilisent des agents d'une classe spécifique de médicaments appelés alpha-bloquants. Ces agents sont utilisés avant l'utilisation de bêta-bloquants pour équilibrer et contrôler au mieux la réponse de la pression artérielle à l'anesthésie.
Dans les rares cas de phéochromocytomes malins et non guéris par chirurgie, une chimiothérapie ou une radiothérapie peut être nécessaire après la chirurgie. Des essais de nouveaux médicaments très spécifiques ou «ciblés» appelés inhibiteurs de la tyrosine kinase se sont révélés prometteurs dans le traitement du phéochromocytome et sont à l'étude dans des essais cliniques. Pour l'instant, le traitement médicamenteux de cette maladie ne peut pas offrir de guérison possible, mais peut être bénéfique pour le patient en réduisant les symptômes et en prolongeant parfois la vie.
Le phéochromocytome est bénin dans la plupart des cas, et si les complications chirurgicales liées à la tension artérielle peuvent être évitées, les chances de guérison sont excellentes. Les phéochromocytomes malins et bénins peuvent réapparaître après la chirurgie. Les statistiques varient d'une étude à l'autre, mais les taux de récidive se situent en moyenne autour de 10 %. Par conséquent, les soins de suivi à long terme après la chirurgie sont très importants pour garder des perspectives acceptables avec des traitements médicaux ou chirurgicaux appropriés supplémentaires.
Dans le faible pourcentage de ces tumeurs déjà rares dans lesquelles un comportement malin est évident, la survie peut encore être assez prolongée, car le rythme de la maladie peut encore être lent. La participation aux essais cliniques de nouvelles thérapies est fortement encouragée dans le cas malheureux du phéochromocytome métastatique. Si un phéochromocytome est diagnostiqué pendant la grossesse, la mortalité (risque de décès) est augmentée pour la mère et le fœtus. Il est recommandé de référer dès que possible à un centre majeur ayant une expérience avec cette circonstance.
 Le régime de santé planétaire :bon pour notre intestin ?
Le régime de santé planétaire :bon pour notre intestin ?
 Soulagement des brûlures d'estomac – Éteignez le feu
Soulagement des brûlures d'estomac – Éteignez le feu
 Objectif de l'endoscopie :diagnostic, traitement et dépistage
Objectif de l'endoscopie :diagnostic, traitement et dépistage
 Salade de tacos costauds (PRENEZ DANS MON VENTRE)
Salade de tacos costauds (PRENEZ DANS MON VENTRE)
 Les personnes présentant des symptômes du SCI sont susceptibles d'avoir de faibles niveaux de vitamine D,
Les personnes présentant des symptômes du SCI sont susceptibles d'avoir de faibles niveaux de vitamine D,
 Nutrition pour la colite microscopique
Nutrition pour la colite microscopique
 La restriction calorique entraîne une perte de poids mais peut favoriser l'apparition de bactéries pathogènes
La façon dont le régime alimentaire affecte le poids dune personne semble être beaucoup plus complexe quon ne le pensait en raison du rôle potentiel que joue le microbiome intestinal dans labsorption
La restriction calorique entraîne une perte de poids mais peut favoriser l'apparition de bactéries pathogènes
La façon dont le régime alimentaire affecte le poids dune personne semble être beaucoup plus complexe quon ne le pensait en raison du rôle potentiel que joue le microbiome intestinal dans labsorption
 TDM d'enfants aux urgences souffrant de douleurs à l'estomac qui montent en flèche
Dernières actualités Enfants en bonne santé Les problèmes dalimentation du bébé liés à des retards de développement Son bras sest coincé dans le tapis roulant de la famille Vous vous inquiétez de lut
TDM d'enfants aux urgences souffrant de douleurs à l'estomac qui montent en flèche
Dernières actualités Enfants en bonne santé Les problèmes dalimentation du bébé liés à des retards de développement Son bras sest coincé dans le tapis roulant de la famille Vous vous inquiétez de lut
 Plus de légumes, de céréales complètes et de diversité de matières grasses, la recette de RD Kate Scarlata pour un microbiote sain
Dans cette vidéo proposée par GMFH, nous avons discuté avec la diététicienne Kate Scarlata, basée à Boston, de la façon dont le microbiote intestinal influence notre santé et notre bien-être. Syndrome
Plus de légumes, de céréales complètes et de diversité de matières grasses, la recette de RD Kate Scarlata pour un microbiote sain
Dans cette vidéo proposée par GMFH, nous avons discuté avec la diététicienne Kate Scarlata, basée à Boston, de la façon dont le microbiote intestinal influence notre santé et notre bien-être. Syndrome