 Jemand mit einem Phäochromozytom hat normalerweise drei klassische Symptome – Kopfschmerzen, Schwitzen und Herzklopfen (schneller Herzschlag) in Verbindung mit deutlich erhöhtem Blutdruck (Hypertonie).
Jemand mit einem Phäochromozytom hat normalerweise drei klassische Symptome – Kopfschmerzen, Schwitzen und Herzklopfen (schneller Herzschlag) in Verbindung mit deutlich erhöhtem Blutdruck (Hypertonie).
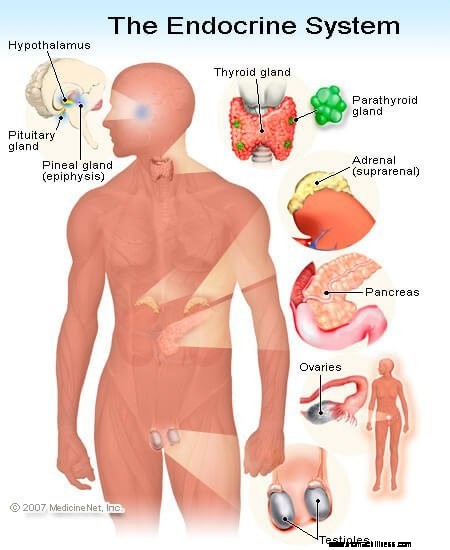
Phäochromozytome sind eine Art Tumor der Nebennieren, die große Mengen an Epinephrin und Norepinephrin freisetzen können. Wie der Name schon sagt, befinden sich die Nebennieren in der Nähe des „Nieren“-Bereichs. Auf jeder der beiden Nieren sitzt eine Nebenniere.
Trotz ihrer geringen Größe haben die Nebennieren viele Funktionen. Sie sind komplexe endokrine (hormonausscheidende) Drüsen. Zellen in verschiedenen Regionen der Nebenniere haben unterschiedliche Funktionen im endokrinen System. Der äußere Teil der Nebenniere wird als Nebennierenrinde bezeichnet. In einem Teil der Nebennierenrinde (Zona fasciculata und Zona reticularis) scheiden die Zellen Cortisol aus, ein Cortison ähnliches Hormon, das für die Stressbewältigung unerlässlich ist. In einem anderen Bereich (Zona glomerulosa) scheiden Zellen ein Hormon namens Aldosteron aus, das bei der Wasser- und Salzregulierung hilft und Blutdruckkontrolle.
Der innere Bereich der Nebenniere wird als Nebennierenmark bezeichnet. Hier scheiden die Zellen Substanzen aus, die Katecholamine genannt werden – Epinephrin, Norepinephrin und Dopamin. Dies sind "Flucht-oder-Kampf"-Hormone. Sie sind zum Teil verantwortlich für das Gefühl eines „Adrenalinschubs“, den Menschen empfinden, wenn sie Angst haben. Es sind diese Zellen, die von einem Phäochromozytom überproduziert werden. Grundsätzlich ist ein Phäochromozytom ein Tumor dieser Katecholamin-sezernierenden Zellen, und das verursacht die klinischen Anzeichen und Symptome, die wir weiter unten besprechen werden. Die Katecholamin-sekretierenden Zellen werden manchmal als chromaffine Zellen bezeichnet und sie werden in anderen Bereichen des Körpers sowie im Nebennierenmark gefunden.
Manchmal entstehen Phäochromozytome aus chromaffinen Zellen, die sich außerhalb der Nebenniere befinden. In diesem Fall werden sie als extraadrenale Phäochromozytome oder Paragangliome bezeichnet und befinden sich normalerweise im Abdomen.
Phäochromozytome können bei Personen jeden Alters auftreten. Der Häufigkeitsgipfel liegt zwischen dem dritten und fünften Lebensjahrzehnt. Phäochromozytome sind glücklicherweise ziemlich selten und die meisten von ihnen sind gutartig.
Bluthochdruck oder Hypertonie tritt am häufigsten ohne Symptome auf und wird aus diesem Grund als „stiller Killer“ bezeichnet. Eine unkomplizierte Hypertonie kann jahrelang, sogar Jahrzehnte bestehen, ohne Symptome zu verursachen. Wenn sich jedoch aufgrund einer Schädigung des Gefäßsystems Komplikationen der Erkrankung entwickeln, können Symptome auftreten.
Erfahren Sie mehr über die Ursachen von Bluthochdruck »
Jemand mit einem Phäochromozytom hat normalerweise drei klassische Symptome – Kopfschmerzen, Schwitzen und Herzklopfen (schneller Herzschlag) in Verbindung mit deutlich erhöhtem Blutdruck (Hypertonie). Andere Zustände, die diese klassischen Symptome begleiten können, sind folgende:Angst, Übelkeit, Zittern, Schwäche, Bauchschmerzen und Gewichtsverlust.
Manche Menschen entwickeln jedoch nie Symptome eines Phäochromozytoms. Bis zu 10 % der Fälle werden zufällig entdeckt, was bedeutet, dass sie nicht vermutet werden und nur gefunden werden, wenn der Patient diagnostischen Studien für andere Erkrankungen unterzogen wird. In einigen Fällen kommt und geht der Bluthochdruck und kann schwierig zu dokumentieren sein. In anderen Fällen ist der Blutdruck konstant erhöht und leicht zu erfassen.
Phäochromozytome kommen nur bei etwa 0,2 % aller Menschen mit Bluthochdruck vor. Es gibt jedoch bestimmte Bedingungen, bei denen die Diagnose eines Phäochromozytoms ganz oben auf der Liste der Möglichkeiten stehen kann; sie werden weiter unten besprochen.
Phäochromozytome können Bestandteil bestimmter familiärer oder genetischer Syndrome sein. Die häufigste familiäre Erkrankung wird als multiple endokrine Neoplasie oder kurz MEN bezeichnet. Zwei Typen von MEN – MEN 2A und 2B – sind mit Phäochromozytomen assoziiert. Beides sind genetische Syndrome, die in Familien vorkommen und autosomal-dominant von den Eltern auf das Kind übertragen werden.
Phäochromozytome sind nicht die einzigen Tumoren, die bei MEN 2A und 2B auftreten. MEN 2A birgt ein erhöhtes Risiko für Tumore der Nebenschilddrüsen, Drüsen in der Nähe der Schilddrüse, die helfen, den Kalziumspiegel im Körper zu regulieren. Und sowohl MEN 2A als auch 2B erhöhen das Risiko eines medullären Schilddrüsenkarzinoms – einer ungewöhnlichen Art von Schilddrüsenkrebs. In Familien mit Verdacht auf MEN können Gentests durchgeführt werden, um gefährdete Familienmitglieder zu identifizieren.
Phäochromozytome sind ein Merkmal anderer genetischer Erkrankungen, einschließlich des von Hippel-Lindau-Syndroms und der Neurofibromatose Typ 1. Diese beiden Erkrankungen sind mit der Entwicklung zahlreicher gutartiger und bösartiger Tumore verbunden.
Es gibt auch viele Personen, die Phäochromozytome ohne bekannte Familienanamnese haben. Diese Fälle werden als sporadisch bezeichnet. Wenn diese Patienten eine bilaterale Erkrankung haben (Phäochromozytome in beiden Nebennieren) oder vor dem 21. Lebensjahr diagnostiziert werden, wird im Allgemeinen ein genetisches Screening empfohlen.
Grundsätzlich kann alles, was zu einer Überaktivität des sympathischen Nervensystems führen kann, auf der Liste der auszuschließenden Diagnosen bei Verdacht auf ein Phäochromozytom stehen. Das sympathische System ist das Hauptsteuerpult, das die Auslösung der „Flucht oder Kampf“-Reaktion als Reaktion auf Stress oder Angst steuert, wie oben erwähnt. Zu den Dingen, die dies stimulieren können, gehören Medikamente (sogar der übermäßige Gebrauch von abschwellenden Mitteln sollte in Betracht gezogen werden); Entzug von Medikamenten (z. B. plötzliches Absetzen bestimmter Blutdruckmedikamente); Panikattacken und Rückenmarksverletzungen gehören zu den vielen Erkrankungen, die auch zu einigen der bei Phäochromozytomen beobachteten Symptome führen können.
Das Phäochromozytom ist bei jedem mit der klassischen Trias von Symptomen – Kopfschmerzen, Schwitzen und Herzklopfen – möglich, insbesondere bei hohem Blutdruck (obwohl hoher Blutdruck nicht immer vorhanden ist). Ein Arzt wird misstrauischer, wenn der Patient jung ist und keine anderen Risikofaktoren oder Gewohnheiten aufweist, die diese Befunde verursachen könnten.
Vielleicht kennt der Patient seine Familienanamnese gut und kann seinen Arzt über andere Arten von endokrinen Tumoren informieren, einschließlich Phäochromozytom(en), die bei Familienmitgliedern diagnostiziert wurden. Folglich kann ein Arzt, der ein familiäres Phäochromozytom vermutet, direkt zu Gentests gehen. In den meisten Fällen führt der Arzt jedoch bei starkem Verdacht eine Reihe von Tests durch, um die unten aufgeführten Hormone und ihre Abbauprodukte (Metaboliten) zu messen.
Zunächst werden Hormone wie Katecholamine und Metanephrine in einer 24-Stunden-Urinsammlung gemessen, und Metanephrine können auch im Blut gemessen werden. Wenn diese mehr als das Zweifache des Normalwertes betragen, werden normalerweise bildgebende Untersuchungen durchgeführt, um die Nebennieren zu untersuchen.
Wenn die Bildgebung mit MRT- oder CT-Scans der Nebennieren eine Masse in der Drüse (oder außerhalb) zeigt, kann eine Operation durchgeführt werden. Wenn nicht klar ist, dass die Raumforderung tatsächlich funktionell ist und klinisch mit den Befunden in Zusammenhang steht, oder wenn in der Bildgebung keine Raumforderung zu sehen ist, kann ein weiterer Test durchgeführt werden. Dieser als 131-I-MIBG-Scan bezeichnete Test ist ziemlich spezifisch für Phäochromozytome. Bei diesem Test wird ein radioaktives Jodmolekül in die Blutbahn injiziert und im Bereich des Tumors lokalisiert, wodurch das Phäochromozytom bei bildgebenden Untersuchungen sichtbar gemacht werden kann.
Die Operation ist die endgültige Behandlung. Bis der Tumor entfernt ist, hat die Kontrolle des Blutdrucks oberste Priorität. Die Kontrolle des Blutdrucks vor und während der Operation ist der schwierigste Teil der Pflege. Es besteht die Möglichkeit, dass eine akute hypertensive Krise (ein potenziell gefährlicher, plötzlicher und schwerer Anstieg des Blutdrucks) entsteht, nachdem während der Operation eine Anästhesie durchgeführt wurde. Der Blutdruck wird daher sowohl vor als auch während der Operation mit speziellen Medikamenten kontrolliert und während des gesamten Eingriffs sorgfältig überwacht. Es wird empfohlen, sich von einem Endokrinologen beraten zu lassen, um eine Behandlung für einzelne Patienten zu entwickeln.
Aufgrund der Art der Hormone, die beim Phäochromozytom beteiligt sind, werden bei ersten Versuchen zur Blutdruckkontrolle Mittel einer bestimmten Klasse von Arzneimitteln verwendet, die als Alpha-Blocker bekannt sind. Diese Wirkstoffe werden vor der Anwendung von Betablockern verwendet, um die Reaktion des Blutdrucks auf die Anästhesie auszugleichen und optimal zu kontrollieren.
In den seltenen Fällen von bösartigen Phäochromozytomen, die nicht durch eine Operation geheilt werden, kann nach der Operation eine Chemotherapie oder Strahlentherapie erforderlich sein. Versuche mit sehr spezifischen oder "zielgerichteten" neuen Medikamenten, die als Tyrosinkinase-Inhibitoren bezeichnet werden, haben sich als vielversprechend bei der Behandlung von Phäochromozytom erwiesen und werden derzeit in klinischen Studien untersucht. Bislang kann die medikamentöse Therapie dieser Krankheit keine mögliche Heilung bieten, kann aber dem Patienten zugute kommen, indem sie die Symptome lindert und manchmal das Leben verlängert.
Das Phäochromozytom ist in den meisten Fällen gutartig, und wenn blutdruckbedingte chirurgische Komplikationen vermieden werden können, sind die Heilungschancen ausgezeichnet. Sowohl maligne als auch gutartige Phäochromozytome können nach der Operation wieder auftreten. Die Statistiken variieren von einer Studie zur nächsten, aber die Rezidivraten liegen im Durchschnitt bei etwa 10 %. Daher ist eine langfristige Nachsorge nach der Operation sehr wichtig, um die Aussichten mit zusätzlichen geeigneten Behandlungen durch Medikamente oder Operationen fair bis gut zu halten.
Bei dem geringen Prozentsatz dieser bereits seltenen Tumoren, bei denen ein bösartiges Verhalten offensichtlich ist, kann das Überleben noch ziemlich verlängert sein, da das Tempo der Krankheit immer noch langsam sein kann. Die Teilnahme an klinischen Studien zu neuen Therapien wird im unglücklichen Fall des metastasierten Phäochromozytoms dringend empfohlen. Wird während der Schwangerschaft ein Phäochromozytom diagnostiziert, ist die Sterblichkeit (Sterblichkeitsrisiko) sowohl für die Mutter als auch für den Fötus erhöht. Eine schnellstmögliche Überweisung an ein größeres Zentrum mit Erfahrung in dieser Situation wird empfohlen.
 Kann ich 2 Tage vor der Darmspiegelung Kartoffelpüree essen?
Kann ich 2 Tage vor der Darmspiegelung Kartoffelpüree essen?
 Was stoppt Durchfall schnell?
Was stoppt Durchfall schnell?
 Ein Überblick über Lymphknoten-positiven Brustkrebs
Ein Überblick über Lymphknoten-positiven Brustkrebs
 Viel rotes Fleisch kann mit Darmerkrankungen bei Männern in Verbindung gebracht werden
Viel rotes Fleisch kann mit Darmerkrankungen bei Männern in Verbindung gebracht werden
 Woher wissen Sie, ob Sie Malabsorption haben?
Woher wissen Sie, ob Sie Malabsorption haben?
 Wir verändern gemeinsam die Welt (ein Bauch nach dem anderen)
Wir verändern gemeinsam die Welt (ein Bauch nach dem anderen)
 Erkrankung der Gallenblase
Die Gallenblase ist eines der kleinen Organe des Magen-Darm-Trakts, über das sich viele Menschen erst dann Gedanken machen, wenn es ein Problem gibt. Tatsächlich spielt die Gallenblase jedoch eine wic
Erkrankung der Gallenblase
Die Gallenblase ist eines der kleinen Organe des Magen-Darm-Trakts, über das sich viele Menschen erst dann Gedanken machen, wenn es ein Problem gibt. Tatsächlich spielt die Gallenblase jedoch eine wic
 Top 10 IBS- und FODMAP-Blogs
Es kann entmutigend sein, glaubwürdige IBS- und niedrige FODMAP-Informationen zu finden. Wir bei Ignite glauben wirklich, dass es keinen Sinn macht, das Rad neu zu erfinden – weshalb wir uns entschlos
Top 10 IBS- und FODMAP-Blogs
Es kann entmutigend sein, glaubwürdige IBS- und niedrige FODMAP-Informationen zu finden. Wir bei Ignite glauben wirklich, dass es keinen Sinn macht, das Rad neu zu erfinden – weshalb wir uns entschlos
 Leberkrebs (hepatozelluläres Karzinom)
Fakten zu Leberkrebs Leberkrebs ist oft das Ergebnis einer chronischen Lebererkrankung. Die meisten Menschen, die Leberkrebs (Leberkrebs) bekommen, bekommen ihn im Rahmen einer chronischen Lebererkra
Leberkrebs (hepatozelluläres Karzinom)
Fakten zu Leberkrebs Leberkrebs ist oft das Ergebnis einer chronischen Lebererkrankung. Die meisten Menschen, die Leberkrebs (Leberkrebs) bekommen, bekommen ihn im Rahmen einer chronischen Lebererkra