celular de cáncer gástrico en todo el genoma número de copias de genes y análisis de la expresión de los tumores gástricos primarios y líneas celulares de cáncer gástrico
Resumen Antecedentes
El cáncer gástrico es una de las neoplasias más comunes en todo el mundo y la segunda causa más común de muerte relacionada con el cáncer. número de copias génicas alteraciones desempeñan un papel importante en el desarrollo de cáncer gástrico y un cambio en el número de copias del gen es uno de los principales mecanismos de una célula de cáncer para controlar la expresión de oncogenes potenciales y genes supresores de tumores.
Métodos
para poner de relieve los genes de potencial relevancia biológica y clínica en el cáncer gástrico, llevamos a cabo un sistemático estudio basado en la serie de la expresión génica y copiamos los niveles numéricos en los tumores gástricos primarios y líneas celulares de cáncer gástrico y validado los resultados utilizando una transcripción análisis basado captura por afinidad ( ensayo de TRAC) y en tiempo real QRT-PCR.
resultados
análisis de microarrays integrado revelaron un total de 256 genes que fueron localizados en regiones recurrentes de las ganancias o pérdidas y que tenían al menos un cambio asociado de 2 veces copia en su número- la expresion genica. Los niveles de expresión de 13 de estos genes, ALPK2
, ASAP1, CEACAM5
, CYP3A4, ENAH
, ErbB2
, HHIPL2
, LTB4R
, MMP9
, PERLD1
, PNMT
, Ptpra
, y OSMR
, fueron validados en un total de 118 muestras gástricas usando ya sea el QRT-PCR o ensayo de TRAC. Todos estos 13 genes son expresados diferencialmente entre las muestras cancerosas y tejidos no malignos (p < 0,05) y la asociación entre el número y cambios de expresión génica de copia fue validado por nueve (69,2%) de estos genes (p < 0,05).
conclusión
En conclusión, la expresión del gen integrado y el análisis de microarrays número de copias de relieve genes que pueden ser de importancia crítica para la carcinogénesis gástrica. TRAC y QRT-PCR análisis validados los resultados de microarrays y por lo tanto el papel de estos genes como marcadores biológicos potenciales para el cáncer gástrico.
Antecedentes
Debido a la falta de síntomas tempranos adenocarcinoma gástrico se caracteriza por la etapa tardía diagnóstico y opciones poco satisfactorios para las tratamiento curativo [1, 2]. A pesar de la disminución de su incidencia en las últimas décadas, el cáncer gástrico sigue siendo la segunda causa más común de muerte por cáncer en todo el mundo [3]. Aproximadamente el 90% de todos los cánceres gástricos son adenocarcinomas que surgen de epitelio [4]. De acuerdo con los cánceres gástricos clasificación de Lauren se dividen en dos principales subtipos histológicos, intestinal y difuso [5].
Adenocarcinomas gástricos, como muchos otros tumores sólidos de origen epitelial, son a menudo complejas en términos de integridad cromosómica [6, 7]. tumores gástricos malignos son conocidos por llevar múltiples aberraciones en su genoma y tales alteraciones cromosómicas son cruciales para la activación e inactivación de los genes relacionados con el cáncer [8-17]. Gen cambio de número de copia es uno de los principales mecanismos para una célula de cáncer para controlar la expresión de genes cruciales para la supervivencia celular y la progresión del cáncer [17-22]. Estas alteraciones del número de copias a menudo implican un gran grupo de genes localizados cerca uno del otro en el mismo cromosoma. Por ejemplo; en los cánceres gástricos la región 17q12-q21 amplificada con frecuencia contiene los genes, como ErbB2
, GRB7
, JUP
, PERLD1, PNMT
, PPP1R1B
, STARD3
, y TOP2A
[14, 17, 23]. Sin embargo, sólo una minoría de estos genes es probable que sean los verdaderos genes del cáncer de controladores que contribuyen a la tumorigénesis, mientras que otros pueden ser amplificados simplemente debido a su cercanía cromosómica con los genes diana de amplificación [24, 25]. Un enfoque para distinguir tales genes controladores de las mutaciones de pasajeros es integrar el número de copias de todo el genoma y los datos de expresión, que permite la identificación de genes cuya transcripción activación o represión está asociada con un cambio de número de copia en una célula de cáncer. De este modo, combinando la información de los números y de expresión de copias de genes microarrays de alta resolución, es posible no sólo para definir los puntos de interrupción de cambios de número de copias en gran detalle, sino también para evaluar la importancia funcional de estos cambios y por lo tanto posiblemente identificar los genes que impulsan el cáncer inicio y la progresión.
a poner de relieve los posibles genes como marcadores biológicos u objetivos clínicos en cáncer gástrico, llevamos a cabo un estudio sistemático basado en matrices de alta resolución del número de copias y la expresión génica en los niveles de cáncer gástrico tejidos y líneas celulares. Nuestro análisis basado en la matriz anterior mostró que las ganancias de número de copia y las pérdidas de cientos de genes están asociados con un aumento simultáneo o disminución de la expresión de genes [17]. En el presente estudio, hemos aumentado la resolución del análisis del número de copia más de 20 veces para visualizar con mayor precisión los puntos de interrupción de las alteraciones del número de copias. Por otra parte, hemos llevado a cabo un análisis transcripcional de los genes localizados en regiones cromosómicas alteradas para identificar los genes cuya desregulación está asociada con el fenotipo maligno.
Métodos
tejidos de cáncer gástrico y líneas celulares
Este proyecto de investigación se ha revisado y aprobado por el Comité ético del Departamento de Genética médica y Cirugía y autorizado por la Junta de Revisión clínica del hospital central Universitario de Helsinki. muestras de tejido gástrico se recogieron de forma prospectiva de pacientes que se sometieron a cirugía gástrica o gastroscopia en el Hospital Central Universitario de Helsinki entre 1999 y 2007. El consentimiento informado se obtuvo de cada paciente que participa. Trece tejidos frescos congelados primarios gástricos cáncer y siete líneas celulares de cáncer gástrico se eligieron para el análisis de microarrays (Tabla 1). El material tejido consistió en dos subtipos histológicos diferentes, intestinal (n = 9) y difusa (n = 4) y los tumores se encuentran en dos sitios diferentes del estómago, el corpus (n = 8) y el antro (n = 5 ). En total, 111 tejidos gástricos y 7 líneas celulares de cáncer gástrico se incluyeron en el QRT-PCR y el ensayo basado en la transcripción de captura por afinidad (TRAC) análisis (archivo adicional 1: Los parámetros clínicos). Las muestras de tejido no maligno consistieron de 43 y 68 tejidos gástricos cancerosas y fueron representados ambos subtipos histológicos de cáncer gástrico (intestinal, n = 42; difusa, n = 25; uno de histología desconocida). muestras de tejido gástrico se almacenaron a -80 ° C. Para verificar el porcentaje del tumor y la histología de las muestras, las muestras congeladas fueron incorporados en Tissue-Tek Compuesto OCT (Sakura Finetek, Torrance, CA, EE.UU.) y 5 micras de hielo secciones congeladas se prepararon y se tiñeron usando azul de tripano. La histología de las muestras de cáncer gástrico fue evaluada por un patólogo experimentado (M.-L. K.-L.). Tissue-Tek se retiró de los tejidos antes de la nucleico extractions.Table 1 Parámetros clínicos de ácido para las muestras analizadas en la matriz de hibridación genómica comparada (aCGH) y la expresión microarrays.
primarios gástrico tumors
Age/sex
Histology
Location
14TA
58/M
Intestinal
Corpus
200A
57/F
Intestinal
Corpus
222A
50/M
Intestinal
Corpus
232A
83/M
Intestinal
Corpus
3TC
57/F
Intestinal
Corpus
4T/N
72/M
Intestinal
Corpus
10TB
59/M
Intestinal
Antrum
17TA
77/M
Intestinal
Antrum
185B
78/F
Intestinal
Antrum
1AT/N
41/F
Diffuse
Corpus
6TB
77/F
Diffuse
Corpus
9TD
74/F
Diffuse
Antrum
13TA
56/F
Diffuse
Antrum
líneas celulares de cáncer gástrico
Edad /sexo
Histología
Origen
AGS
54 /F
adeno-carcinoma de tumor primario
KATOIII
55 /M
derrame pleural difusa
MKN-1 | 72 /M
carcinoma adeno-escamosas
metástasis de ganglios linfáticos
MKN-7
39 /M
intestinal
metástasis de ganglios linfáticos
MKN-28
70 /F
intestinal
metástasis de ganglios linfáticos
MKN-45
62 /F
difusa
metástasis de hígado
TMK-1 | 21 /M
difuso de metástasis en los ganglios linfático
líneas de células AGS y KATOIII se obtuvieron de la American Type Culture Collection (Rockville, MD, EE.UU.) y MKN-1, MKN-7 , MKN-28, MKN-45, y las líneas celulares TMK-1 fueron una especie de regalo de Hiroshi Yokozaki, Kobe University Graduate School of Medicine, Kobe, Japón [26]. células AGS se cultivaron en medio F12 de Kaighn (glutamina 2 mM, 10% de SFB, 100 U /ml de penicilina-estreptomicina), las células KATOIII en medio IMDM (glutamina 2 mM, 10% de SFB, 100 U /ml de penicilina-estreptomicina) y todos otras líneas celulares en medio RPMI-1640 (10% de FCS, glutamina 2 mM, 100 U /ml de penicilina-estreptomicina). Todas las células se cultivaron a 37 ° C y 5% de CO
2.
ARN y la extracción de ADN
Antes de ARN y ADN extracciones, el tejido congelado se sumergió en reactivo RNAlater-ICE (Ambion, Austin, TX , EE.UU.) y almacenados a -80 ° C durante 16 horas para estabilizar el ARN. La mitad de la muestra de tejido (~ 25 mg) se homogeneizó en tampón RLT-β-mercaptoetanol lisis (RNeasy mini kit, Qiagen Inc., Hilden, Alemania) y la otra mitad en ATL-buffer (DNeasy Tissue Kit de la sangre y, Qiagen) usando el homogeneizador Ultra-Turrax (IKA Works, Wilmington, NC, EE.UU.). ARN fue extraído utilizando el RNeasy mini kit, incluyendo el tratamiento con DNasa opcional, y el ADN utilizando la sangre y DNeasy Tissue Kit. Para las líneas celulares de cáncer gástrico, 1 × 10 7 células se lisaron usando una jeringa y la aguja en tampón de lisis, ya sea RLT-β-mercaptoetanol o ATL-buffer antes de ARN y ADN extracciones, respectivamente. Las concentraciones de ARN y ADN se midieron utilizando NanoDrop1000 (Thermo Fisher Scientific, Waltham, MA, EE.UU.) y se evaluó la calidad del ARN usando de Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, EE.UU.). Sólo los ARN que muestran distintas 18S y 28S ribosomal picos en el análisis Bioanalyzer y 260/280 relaciones por encima de 2,0 se aceptaron para el análisis adicional. Analiza
matriz CGH y microarrays de expresión génica
se analizaron trece tumores gástricos y siete líneas celulares de cáncer gástrico en los oligoarrays 244K Genoma humano CGH (G4411B, Agilent Technologies). Tres de los tumores y todas las siete líneas celulares también fueron analizados utilizando el 44K de todo el genoma humano de expresión génica oligoarrays (G4112F, Agilent Technologies) (Figura 1). La media de 260/280 relaciones para estas muestras fueron 2,1 para el ARN y 1,8 para el ADN, y todas las muestras de ARN tenido claros picos de 18S y 28S ribosomal en el análisis Bioanalyzer que indica buena calidad (datos no mostrados). CGH array experimentos se llevaron a cabo utilizando el kit del Genoma Humano Microarray CGH 244A (Agilent Technologies). Etiquetado y la hibridación se realizaron de acuerdo con el protocolo del Agilent (v5.0, junio de 2007). En resumen, 1,5 mg de ADN de la muestra y 1,5 g de ADN de referencia de sexo-emparejados (ADN genómico humano, Promega, Madison, WI, EE.UU.) fueron doble digerido con AluI y RsaI
enzimas de restricción (Promega). El ADN digerido se marcó utilizando el ADN genómico Agilent Labeling Kit Plus. La muestra de ADN se marcó con Cy5-dUTP y de DNA de referencia con Cy3-dUTP, respectivamente. ADN marcado se purificó con Microcon YM-30 filtros (Millipore, Billerica, MA, EE.UU.). Después de los ADN de purificación, muestra y de referencia se agruparon y se hibridó a la matriz con 50 g de Cot-1 DNA humano (Invitrogen, Carlsbad, CA, EE.UU.) a 65 ° C, 20 rpm durante 40 h. La hibridación se realizó con el Kit de Agilent Oligo aCGH hibridación. Antes de la digitalización, los portaobjetos se lavaron de acuerdo con el protocolo. Además de las hibridaciones de ADN de muestra descritos anteriormente, varón de ADN de referencia (Cy3) se hibridó contra ADN femenino de referencia (Cy5) de acuerdo con el mismo protocolo para ser utilizado como una matriz de referencia en el análisis de datos aCGH. Figura 1 Diagrama de flujo que describe las diferentes etapas del estudio.
Gene experimentos de expresión se realizaron utilizando el kit del Genoma Humano Oligo Microarray Total (Agilent Technologies), y el etiquetado y la hibridación según el protocolo de Agilent (v5.7, marzo de 2008). En resumen, 2 g de ARN total de la muestra y el ARN de referencia (un grupo de líneas celulares de cáncer de 10, no gástrico, ATCC, Manassas, MA, EE.UU.) se marcaron usando el Amp Labeling Kit Agilent rápida. ARN de muestra se marcó con Cy5-dCTP y de DNA de referencia con Cy3-dCTP, respectivamente. a continuación, ARN marcado se purificó usando RNeasy mini columnas de centrifugación (Qiagen). La hibridación se realizó con Agilent gen KIT Expresión La hibridación y se hibridaron muestras a 65 ° C, 10 rpm durante 17 h y se lavó según el protocolo antes de la digitalización. Ambos diapositivas de microarrays de expresión génica y aCGH fueron escaneados utilizando el escáner de microarrays de ADN (Agilent Technologies) y se analizaron con el software de extracción de características (v9.5.1.1.).
Número de copias de alta resolución perfilado MyBestPlay Todos los datos de número de copias es disponible en http: //www. cangem org (número de acceso: CG-EXP-49). [27]. Se aplicó el software de análisis de CGH de Agilent (v3.5.14) para identificar los cambios de número de copias. datos de microarrays se filtró calidad usando la información de valor atípico obtenido a partir del análisis de extracción de características. Las sondas marcadas como valores atípicos se eliminaron del análisis adicional. Además, se aplicaron los siguientes filtros de aberración: número mínimo de sondas en la región = 3, registro mínimo absoluto relación 2 para la región = 0,27, y el número máximo de regiones aberrantes = 1000. El registro de la relación 2 de 0,27 corresponde a un cambio de 1,2 veces en el número de copias. En CGH Analytics, cada relación de aCGH se convierte primero en un registro de relación 2 y una Z-normalización. El macho vs. matriz de referencia hembra se utilizó como una matriz de calibración en el análisis de datos. Debido a las diferencias de género entre las matrices que podrían causar sesgo en el análisis, los cromosomas X e Y se excluyeron de la calibración. se utilizó algoritmo de ADM-2 con un nivel de umbral de 12,0 para identificar copias de genes alteraciones en el número de muestras individuales y líneas celulares. Se calcularon las regiones comunes mínimas de alteraciones en las 20 muestras, incluyendo la posición y tamaño cromosómico de la alteración en pares de bases. Una aberración que se definió como recurrente, si estaba presente en al menos el 25% de las muestras (Tabla 2) .Tabla 2 regiones comunes mínimas de recurrente (≥25%) alteraciones en el número de copia.
Alteración
tejidos (n = 13)
líneas celulares (n = 7) guía empresas de frecuencia
Tamaño (Mb) guía empresas Posición (Mb) guía empresas posibles genes diana
+ 1q41-q43.1
2 3 25
17.30%
216,31-233,61
ENAH, AGT, CAPN2, LEFTY2, LGALS8
+ 5p13.3-Q11.1
1
4 de 25%
19.41
30,18-49,60
OSMR, RNASEN
+ 7q21.3-q22.1 página 4 página 3
35%
4,60
97,33 a 101,93
CYP3A4, AZGP1, VGF
+ 8q24.13-q24.3 página 3
2 25%
19,8
126,45 a 146,25
ASAP1, BAI1, KHDRBS3
+ 8q24.3 página 6 página 3
45%
2.23
143,59-145,82
GML, LYPD, AK3
+ 14q11.2
0 página 5
25%
1.05
22,89-23,94
LTB4R
+ 17q12-q21.1 página 3 página 3
30%
0,28
35,02-35,30
erbB2, PPP1R1B, PERLD1, PNMT
+ 17q22-q24.2
2 3
25%
13.65
50,45-64,10
AXIN2, RNF43
+ 19q12-qter página 4 página 3
35%
29.36
33,89-63,25
CEACAM5, APOC1, APOE, CEACAM7, FTL, FUT1, GPR4, HPN, KCNN4, KLK1, KLK12, LYPD3, NLRP7, CCNE1
+ 20p13-qter página 5
3
40%
57.94
0,04-57,98
Ptpra, BLCAP, CD40, CHGB, CST3, Eya2, PI3, ID1, MMP9, BMP-7
-9p24.3-p21 0.1 página 3
4 de 35%
27,81
1,05-28,86
MTAP, CD274, INSL4, JAK2, MLANA, SMARC2, TUSC1
-18q12. 3-q22.2 página 3 página 5
40%
26.11
39,48-65,59
SMAD7, SERPINB2 /B3 /B4 /B5
-18q22.3 -qter
2 5
35%
3,69
70,95-74,65
TSHZ1
-21q11.2-q21.1 página 3
3
30%
4,07
14,37-19,44
HSPA13
-Xq28
4 de 1 | 25%
1,21
152.24- 153,45 Perfil - Número de casos, el tamaño de las regiones comunes mínimas (Mb), y la posición cromosómica de la alteración (Mb) se indican, así como posibles genes diana. regiones de la CNV no se muestran en la tabla. . El aumento de número de copias (+), la pérdida de número de copias (-)
génica análisis de microarrays de expresión MyBestPlay Todos los datos de expresión génica está disponible en http: //www cangem org (número de acceso: CG-EXP.. -49) [27]. Microarray resultados fueron filtradas utilizando valores atípicos calidad definidos por el software de extracción de características y normalizaron de acuerdo con el método de loess, que estaba incluido en el paquete de software. El análisis de la expresión de genes se limita a los genes localizados en las regiones cromosómicas recurrentes con aberraciones (Tabla 2). El objetivo de este enfoque era para resaltar los cambios de expresión génica que se asociaron con cambios en el número de copias de genes, y por lo tanto podría representar oncogenes potenciales o genes supresores de tumores con un papel funcional en el cáncer. En primer lugar, una mediana relación log10 expresión se calculó para todas las sondas dirigidas a un mismo gen. Luego, en dos análisis separados para las ganancias y pérdidas, el nivel medio de expresión de cada gen se comparó entre las muestras con número de copias ganancia /pérdida y muestras con número de copia normal para evaluar el efecto de las alteraciones del número de copias en la expresión génica. La expresión de genes de plegado cambios (FC) se calcularon ya sea dividiendo la mediana de expresión de las muestras cancerosas por la mediana de expresión de las muestras no malignas o dividiendo la expresión mediana de las muestras de cáncer con alteraciones del número de copias (G1) por la expresión media de las muestras de cáncer con número de copia normal (G0). Al menos se consideró significativo un cambio de 2 veces el número de copias asociado en la expresión génica. Sobre la base de estos datos, 13 genes ALPK2
, ASAP1
, CEACAM5
, CYP3A4
, ENAH
, ErbB2, HHIPL2, LTB4R
, MMP9, OSMR
, PERLD1
, PNMT
, y Ptpra
, fueron elegidos para ser validado aún más con el análisis de QRT-PCR y TRAC (transcripción análisis con ayuda de captura por afinidad) de ensayo. Los resultados de los análisis de microarrays integrado se compararon con tres estudios publicados anteriormente que se integran de forma sistemática el número de copias y la expresión génica de datos de todo el genoma [15-17]. En tiempo real QRT-PCR análisis
en tiempo real QRT PCR se realizó durante 2 genes, ALPK2
(18q21.31-q21.32) y HHIPL2
(1q41). Los niveles de expresión se midieron en 82 tejidos gástricos (46 cancerosa y 36 tejidos no malignos) y en 7 líneas celulares de cáncer gástrico (archivo adicional 1: Los parámetros clínicos). 1 g de RNA total se convirtió en cDNA usando virus de la leucemia Moloney murina-transcriptasa inversa (Promega, Madison, WI, EE.UU.) y cebadores aleatorios (Invitrogen) en un volumen de 50 l para 1 h a 37 ° C. La reacción fue inactivada por calor (95 ° C, 3 min) y llenado a un volumen final de 200 l con agua de grado molecular. Las transcripciones se cuantificaron utilizando los productos de expresión de genes ensayos-on-DemandTM (Hs01085414_m1 para ALPK2 Opiniones y Hs00226924_m1 para HHIPL2
) de acuerdo con el protocolo del fabricante (Applied Biosystems, Foster City, CA, EE.UU.). Todos los cebadores se encuentran en los límites exón-exón. Brevemente, 2 l de molde de ADNc se mezcló con 1,25 l de cebadores y sondas específicas marcadas con colorante FAM-reporter. Se añadieron 12,5 l de TaqMan ® PCR Universal Mastermix y RNasa libre de agua hasta un volumen total de 25 l. Human 18S rRNA sirvió como control endógeno para normalizar los niveles de expresión en el análisis cuantitativo posterior. La sonda 18S fue marcado con colorante VIC-reportero para permitir PCR múltiple con los genes diana. Las condiciones de PCR fueron las siguientes: 50 ° C durante 2 min, 95 ° C durante 10 min, seguido de 40 ciclos de 95 ° C durante 15 s y 60 ° C durante 1 min. Cada muestra se midió por triplicado y los datos se analizaron mediante el método delta-delta para comparar los resultados relativos de expresión (2 - [Ct control de la muestra-Ct]) ensayo de TRAC
transcripción análisis con ayuda de. captura por afinidad (TRAC) de ensayo [28] se llevó a cabo durante 11 genes diferentes en 88 tejidos gástricos (53 canceroso y 35 tejidos no malignos) y 7 líneas celulares de cáncer gástrico (archivo adicional 1: Los parámetros clínicos). Los genes incluidos en el análisis fueron ENAH gratis (1q42.12), OSMR gratis (5p13.1), CYP3A4 gratis (7q21.1) ASAP1 gratis (8q24.1-q24.2), LTB4R gratis (14q11.2-q12), PERLD1 gratis (17q12), erbB2 gratis (17q21.1), PNMT gratis (17q21-q22), CEACAM5 gratis (19q13.1-q13 0.2), Ptpra gratis (20p13), y MMP9 gratis (20q11.2-q13.1). La ventaja del ensayo de TRAC es que los niveles de expresión de múltiples genes se pueden medir simultáneamente de una sola muestra reduciendo así la cantidad de ARN de muestra necesario para el análisis. Esto es especialmente importante para el análisis de frecuencia escasos muestras de tejidos clínicos. Análisis TRAC
se realizó a PlexPress (Helsinki, Finlandia). reactivos TRACPackTM personalizada para mRNA (PlexPress) se utilizaron en el análisis. (Sondas que contienen etiquetados específicos de gen de detección y sondas de oligo-dT biotinilados) Brevemente, 90 l de hibridación Mix por pocillo se dispensó a una placa de 96 pocillos PCR. Dos microgramos de muestra de ARN se aplicó a cada pocillo en un volumen de reacción total de 100 l. Se añadió una cantidad igual (30 amol /reacción) de control único de hibridación de oligonucleótido sintético 62-mer trenzado, incluyendo una cola poli-A, a cada muestra antes de la hibridación. La hibridación se realizó a 60 ° C, 650 rpm durante 120 minutos (Thermomixer Comfort, Eppendorf, Hamburgo, Alemania). Después de la captura por afinidad de hibridación, purificación, y la elución se realiza utilizando el martín pescador Flex (Thermo Fisher Scientific, Vantaa, Finlandia) procesador de partículas magnéticas. Se añadieron perlas de TRACPACK ™ magnéticas de estreptavidina acoplada (50 g, PlexPress) a la mezcla de hibridación y se permite que se una al ARNm biotinilado-sonda-oligo (dT) -hybrids durante 30 minutos, después de lo cual las perlas se lavaron 5 veces con lavado tampón para eliminar cualquier material no unido. sondas de ARN específicas marcadas se eluyeron con tampón de elución y se detectaron por electroforesis capilar, usando el secuenciador ABI3100 (Applied Biosystems, Cheshire, Reino Unido). Los datos fueron analizados utilizando el software TRACParser (PlexPress).
El análisis estadístico de los datos de QRT-PCR
Una prueba de Mann-Whitney no paramétrico para dos muestras independientes se aplicó para determinar la significación estadística de las diferencias en la expresión de mRNA relativa niveles de ALPK2
y HHIPL2
en muestras gástricas no malignas y cancerosas, así como en muestras de cáncer gástrico de diferentes subtipos histológicos o TNM-etapas. Un valor de p < 0,05 fue considerado estadísticamente significativo (SPSS 17.0). Además, en los dos análisis separados para las ganancias y pérdidas, los niveles de expresión en muestras de cáncer con el número de copias ganancias o pérdidas (G1) se compararon con muestras de cáncer con número de copia normal (G0) para evaluar la asociación entre el número de copias y la expresión génica. se disponía de datos de número de copia para 37 de las muestras gástricas incluidas en el análisis QRT-PCR (archivo adicional 1: Los parámetros clínicos). cambios de expresión génica de plegado se calcula dividiendo la media de expresión de un grupo (por ejemplo, muestras de cáncer) por la media de expresión del otro grupo (por ejemplo, las muestras no malignas).
El análisis estadístico de los datos del ensayo de TRAC
Un control de la hibridación sintética se utilizó en la normalización de datos para eliminar cualquier variación no biológico en los datos. Para cada objetivo, la señal intensidades relativas a esta hibridación de control interno se calcularon. Para los nueve muestras de tejidos analizados en Duplicado Media se utilizó la intensidad de la señal. Se aplicó una prueba de Mann-Whitney no paramétrico para dos muestras independientes para determinar la significación estadística de las diferencias en los niveles relativos de expresión de ARNm de ASAP1
, CEACAM5
, CYP3A4
, ENAH
, ErbB2, LTB4R
, MMP9, OSMR
, PERLD1
, PNMT
, y Ptpra
en muestras gástricas no malignas y cancerosas, así como en muestras de cáncer gástrico de diferentes subtipos histológicos o TNM-etapas. Un valor de p < 0,05 fue considerado estadísticamente significativo (SPSS 17.0). La comparación de los niveles de expresión de genes en muestras con y sin alteraciones del número de copias se realizó como se ha descrito antes para el análisis de QRT-PCR. . Estaban disponibles para 43 muestras de cáncer gástrico datos de número de copia incluida en el análisis del ensayo TRAC
Resultados
número de copias de genes aberraciones
Todas número de copias del gen cambia en muestras individuales se muestran en el archivo adicional 2: número de copias cambios detectado mediante análisis aCGH. regiones mínimas comunes de recurrente (≥25%) alteraciones, así como su tamaño, la frecuencia, los posibles genes diana, y la posición cromosómica en pares de bases se muestran en la Tabla 2. El recurrente ganó regiones se encuentran en 1q41-q43.1 (25% ), 5p13.3-Q11.1 (25%), 7q21.3-q22.1 (35%), 8q24.13-q24.3 (25%), 8q24.3 (45%), 14q11.2 ( 25%), 17q12-q21.1 (30%), 17q22-q24.2 (25%), 19q12-qter (35%), y 20p13-qter (40%). Las regiones recurrentes borrado se encuentra en 9p24.3-p21.1 (25%), 18q12.3-q22.2 (40%), 18q22.3-qter (35%), 21q11.2-q21.1 (30 %), y Xq28 (25%). Todos los cambios de número de copia recurrentes fueron detectables tanto en los cánceres gástricos primarios y en líneas celulares de cáncer gástrico, excepto por el 14q11.2, que se altera sólo en cinco líneas celulares.
Número de copia expresión de genes asociados cambia
En total 256 individuo genes (10% de todos los genes localizados en las regiones cromosómicas recurrentes con alteraciones del número de copias) mostraron al menos un cambio asociado de 2 veces el número de copias en su expresión (intervalo de 2,0 - el 34,6, mediana 3,8) (archivo adicional 3: número asociado gen de copia los cambios de expresión). 226 de estos genes se sobreexpresa y están situados en regiones recurrentes de las ganancias de número de copias, mientras que 30 genes fueron underexpressed y ubicados en las regiones de las pérdidas recurrentes de número de copias. veces el cambio en la expresión de genes se calculó comparando los niveles de expresión de las muestras con alteraciones del número de copias de muestras con número de copias normal en un gen dado. Por lo tanto, un factor de cambio positivo se refiere a un aumento relacionado con el aumento de número de copias en la expresión génica, mientras que un factor de cambio negativo se refiere a una disminución relacionada con la pérdida de número de copias en la expresión génica.
HHIPL2 gratis (Hhip 2 similar), el gen amplificado en la región 1q41-q43.1, mostró la más alta sobreexpresión asociado ganancia del número de copias en el cáncer gástrico de acuerdo con el análisis de microarrays integrado (FC = 26,9). En general, la expresión de genes más alta veces los cambios entre las muestras de cáncer con y sin aumentos del número de copias se detectaron en la región 19q ya que de los 40 genes que muestran > cambios de 5 veces el número de copias asociado en su expresión, 19 (47,5%) se localizaron en la región 19q (archivo adicional 3: Copia de expresión génica número asociado cambios). El gen más underexpressed en las regiones de las pérdidas recurrentes número de copias fue ALPK2 gratis (alfa-quinasa 2) (FC = -34,6) situado en 18q12.3-q22.2.
Anteriormente, tres estudios realizados por nosotros y otros se han publicado que incorpore en forma sistemática el número de copias y la expresión génica de datos de todo el genoma para identificar los genes cuya expresión ha cambiado debido a una serie alteración de la copia en cáncer gástrico [15-17]. La comparación de los genes superpuestos entre estos estudios y el estudio actual reveló 20 genes TOMM20 gratis (1q42.3), GGPS1
(1q43), CYP3A4
(7q21.1), MTAP gratis (9q21 0.3), ASAP1 gratis (8q24.1-q24.2), PPP1R1B gratis (17q12), erbB2 gratis (17q12-q21), SERPINB3 gratis (18q21.3), SERPINB8
(18q21.3), WDR7 gratis (18q21.2-q22), HIF3A gratis (19q13.32), ZNF480 gratis (19q13.33), IL4I1 gratis (19q13.3-q13.4 ), CST3 gratis (20p11.21), Ptpra gratis (20p13), SLC13A3 gratis (20q12-q13.1), DDX27 gratis (20q13.13), PARD6B gratis (20q13.13 ), SGK2 gratis (20q13.2), y TUBB1 gratis (20q13.32) que, o bien se ganó y sobreexpresada o se elimina y underexpressed en nuestro estudio y en al menos uno de los estudios publicados anteriormente. publicado previamente los datos junto con los resultados actuales proporcionan una prueba más de la función biológica de estos genes en el cáncer gástrico.
Validación de los posibles genes diana de cáncer gástrico
en tiempo real QRT-PCR análisis mostró que la expresión de HHIPL2
fue 7,4 veces mayor en las muestras de cáncer gástrico en comparación con los tejidos gástricos no malignas (p < 0,05). Además, la sobreexpresión de HHILP2
se asoció significativamente con el aumento de número de copias (p < 0,05) como la expresión de HHIPL2
era 17,4 veces mayor en las muestras de cáncer con el aumento de número de copias de HHIPL2 gratis (G1 ) que en las muestras de cáncer con número de copia normal de este gen (G0) (Tablas 3 y 4). Según el análisis de QRT-PCR que había una subexpresión 2,9 veces de ALPK2
en cánceres gástricos con pérdidas número de copias (G1) en comparación con los cánceres gástricos con número de copia normal de ALPK2
(g0) (P < 0,05 ). Sorprendentemente, sin embargo, la expresión de ALPK2
en los cánceres gástricos en general era 1,9 veces mayor (p < 0,05) que en los tejidos gástricos no malignas (Tablas 3 y 4). subtipo histológico o TNM-etapa no tuvo un efecto estadísticamente significativo en la expresión de HHIPL2
o ALPK2 gratis (Tabla 3) .Tabla 3 Resultados de la prueba no paramétrica de Mann-Whitney para el análisis de datos TRAC QRT-PCR y (SPSS17.0).
gene
cromosoma
cáncer vs. no maligno
intestinal difusa vs
G1 vs g0
M0 M1 vs
T1-2 T3-4 frente
N0 vs. N1-3
ALPK2
18q21.31-q21.32
p < 0.05
p = 0,104
p Hotel < 0.05
p = 0,451
p = 0,072
p = 0,378
ASAP1
8q24.1-q24.2
p < 0,001
p = 0,319
p = 0,396
p = 0,208
p = 0,232
p = 0,289
CEACAM5
19q13.1-q13.2
p <
 La dieta occidental puede aumentar el riesgo de "sepsis mortal",
La dieta occidental puede aumentar el riesgo de "sepsis mortal",
 La presencia de ciertas bacterias intestinales en las madres podría proteger a los bebés de las alergias alimentarias
La presencia de ciertas bacterias intestinales en las madres podría proteger a los bebés de las alergias alimentarias
 Una dieta vegana podría estimular los microbios intestinales que ayudan a perder peso
Una dieta vegana podría estimular los microbios intestinales que ayudan a perder peso
 La secuenciación de ARN ofrece nuevos conocimientos sobre el microbioma
La secuenciación de ARN ofrece nuevos conocimientos sobre el microbioma
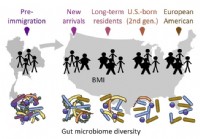 La migración afecta la microbiota intestinal, lo que a su vez afecta la salud encuentran investigadores
La migración afecta la microbiota intestinal, lo que a su vez afecta la salud encuentran investigadores
 Los probióticos pueden ayudar a frenar la desnutrición en las próximas dos décadas,
Los probióticos pueden ayudar a frenar la desnutrición en las próximas dos décadas,
 La apoptosis es un mediador importante de la patogénesis en la infección por coronavirus animal
La apoptosis es una forma de muerte celular programada, que ocurre en células que están dañadas irreparablemente o en células infectadas por algunos virus. Este mecanismo es extremadamente importante
La apoptosis es un mediador importante de la patogénesis en la infección por coronavirus animal
La apoptosis es una forma de muerte celular programada, que ocurre en células que están dañadas irreparablemente o en células infectadas por algunos virus. Este mecanismo es extremadamente importante
 El pH ácido mejora la infección por SARS-CoV-2 regulando positivamente el receptor ACE2
La pandemia actual de la enfermedad por coronavirus 2019 (COVID-19) causada por un nuevo coronavirus, a saber, síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2), ha cobrado más de 4,6 mill
El pH ácido mejora la infección por SARS-CoV-2 regulando positivamente el receptor ACE2
La pandemia actual de la enfermedad por coronavirus 2019 (COVID-19) causada por un nuevo coronavirus, a saber, síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2), ha cobrado más de 4,6 mill