Résumé
Contexte
Alors que les microARN (miARN) jouent un rôle important dans la différenciation des tissus et dans le maintien de la physiologie de base, on sait peu sur les niveaux d'expression de miARN dans les tissus de l'estomac. Les modifications du profil miRNA peuvent conduire à une dérégulation des cellules, ce qui peut induire une néoplasie.
Une petite bibliothèque d'ARN du tissu de l'estomac a été séquence en utilisant à haut débit la technologie de séquençage SOLiD. Nous avons obtenu 261.274 qualité lit avec des allumettes parfaites à la miRnome humaine, et 42% des miARN connus ont été identifiés. Gene Expression numérique profilage (DGE) a été réalisée sur la base de lecture abondance et a montré que quinze miARN ont été fortement exprimés dans le tissu gastrique. Par la suite, l'expression de ces miARN a été validée en 10 individus en bonne santé par RT-PCR a montré une corrélation significative de 83,97% (p < 0,05). Six miARN ont montré une faible tendance variable d'expression (miR-29b, miR-29c, miR-19b, miR-31, miR-148a, miR-451) et pourraient être considérés comme faisant partie du motif d'expression du tissu gastrique sain.
Conclusions /importance
Cette étude visait à valider les profils miRNA normaux de tissu gastrique humaine pour établir un profil de référence pour les personnes en bonne santé. Déterminer les processus réglementaires agissant dans l'estomac sera important dans la lutte contre le cancer de l'estomac, qui est la deuxième cause de mortalité par cancer dans le monde entier
Citation:. Ribeiro-dos-Santos Â, Khayat AS, Silva A , Alencar DO, Lobato J, Luz L, et al. (2010) Le séquençage ultra-profond révèle le motif microRNA d'expression de l'estomac humain. PLoS ONE 5 (10): e13205. doi: 10.1371 /journal.pone.0013205
Editeur: Patrick Tan, Duke-National University de Singapour Graduate Medical School de Singapour
Reçu 30 Mars 2010; Accepté: 8 Septembre 2010; Publié 8 Octobre, 2010
Droit d'auteur: © 2010 Ribeiro-dos-Santos et al. Ceci est un article en accès libre distribué sous les termes de la licence Creative Commons Attribution, qui permet une utilisation sans restriction, la distribution et la reproduction sur tout support, à condition que l'auteur et la source originelle sont crédités
Financement:. Le travail a été soutenue par le Paraense projet Genoma de Genomica e proteómica (Governo do Para /SEDECT /FAPESPA), PROPESP /UFPA, FADESP et CAPES (Coordenação de Aperfeicoamento de Pessoal de Nivel Superior). Les bailleurs de fonds ont joué aucun rôle dans la conception de l'étude, la collecte et l'analyse des données, la décision de publier, ou de la préparation du manuscrit
Intérêts concurrents:.. Les auteurs ont déclaré aucun conflit d'intérêts existent
Introduction
récemment, Friedman et al. Les mécanismes qui impliquent miARN comme régulateurs négatifs de l'expression des gènes sont similaires chez les animaux et les plantes. ils régulent les processus cellulaires fondamentaux [6]. Chez l'homme, environ 3% de tous les gènes sont prédits pour coder des précurseurs de miARN, et plus > 60% des gènes codant pour des protéines pourrait être régulée par miARN [1]. MiRNAs possèdent des fonctions essentielles dans de nombreux processus cellulaires tels que la croissance et le développement, la prolifération cellulaire, la différenciation et l'apoptose. Par conséquent, les modifications de l'expression des miARN contribuent aux maladies humaines telles que les cancers [7], [8]. Des altérations dans les profils d'expression de miARN ont été observées dans de nombreux types de cancer humain, tels que le sein, du côlon, du poumon, la prostate, la leucémie et les cancers gastriques. modifications d'expression de miARN conduisent à une perte ou gain de fonction et sont associés à la mise au point d'un néoplasme humain par l'intermédiaire de divers mécanismes [5]. Ici, nous présentons une étude génétique du miARN de l'estomac humain, parce que, malgré l'importance de l'organe, peu de données génétiques sont disponibles sur leur présence et de la réglementation chez les humains. Ce travail est le premier séquençage miRnome complète des tissus de l'estomac normale en utilisant la technologie de séquençage de nouvelle génération. Dans la présente étude, les profils ont été obtenus par séquençage ultra-profond en utilisant la plate-forme solide ( Life Technologies, CA, États-Unis). Cette étude est la première à utiliser cette procédure pour décrire le miRnome du tissu gastrique normale. miARN ont été isolées de la muqueuse gastrique normale du cardia d'un seul patient sain. Les profils ont été validés à l'aide PCR en temps réel pour déterminer l'expression des 15 miARN les plus fortement exprimés dans 10 patients en bonne santé. Ultra-profonde séquençage a donné un total de 104 millions lectures brutes et 5 millions lit pour la bibliothèque miRNA cardia gastrique. Pour une analyse plus approfondie, 3,2 millions de lectures ont été sélectionnés en fonction de la qualité de la séquence (au moins de QV≥10 dans les 10 premières bases) [8]. count Après avoir cartographié ces au génome humain (libération 19), le total mappée lu était 2.534.490 millions lit (75% de la qualité totale lit). Environ 38% des 2,5 millions de lit (963460 lectures) ont été séquences d'ADN répétitives, ARNt, ARNr ou d'autres petites molécules; 10% (261.274) ont été acceptées lit, parfaitement aligné avec miARN matures connus (version miRBase 15, libérer 04/2010) [9]; et le reste du lit (52%) ont été appariés à génome séquences (figure 1). Pour l'analyse de l'expression des miARN, lit uniquement avec des allumettes à maturité miARN séquences (261.274 lectures) ont été inclus. Dans cardia gastrique, nous avons identifié 404 des 970 séquences matures miARN connus (42%). Pour analyser l'expression des miARN, nous avons défini cinq gammes: 1 à 10 chefs d'accusation de lecture; 11 à 100; 101 à 1000; 1.001 à 5.000; et un nombre de lectures de plus de 5000 (figure 2) (détails supplémentaires sont fournis dans le tableau S1). La première gamme (1-10) comprend 40% des miARN observées; la deuxième plage (11 à 100) composé de 26%; la troisième plage (101 à 1000) composé de 20%; et les quatrième et cinquième rangs (> 1000 comptage de lecture) représentaient 14% Avec cette classification, 347 miARN matures ont été exprimés entre les premier et troisième rangs et 57 entre les quatrième et cinquième rangs.. Pour la caractérisation de la miRnome, nous avons sélectionné un ensemble de 15 miARN qui ont été exprimées au plus haut niveau (≥1,000 lectures). Tableau 1 et Figure 3 Liste des 15 miARN matures identifiées dans les cardia gastriques humaines . Le heatmap de la figure 4 résume l'expression de ces 15 miARN dans le cardia gastrique et dans les tissus humains normaux, des données DGE, disponibles dans la banque de données microRNA.org [10], [11]. expression miRNA mature pourrait être classé en deux groupes: i) cardia-tissus: miARN rarement exprimées dans d'autres tissus, mais exprimée en cardia gastrique, y compris miR-148a, miR-192, miR-200a et miR-200b; ii) quasi-omniprésente: miARN exprimé dans de nombreux tissus et conditions, y compris miR-29c, miR-21, miR-24, miR-29b, miR-29a, miR-451, miR-31, miR-145, miR-26a , miR-19b et laissez-7b. REAL TIME les résultats de séquençage ultra-profonds ont été validés à l'aide du singleplex PCR en temps réel (RT-PCR) sur les 15 miARN sélectionnés déterminer leur expression dans la région du cardia gastrique chez 10 individus sains. Parmi les sujets de test, 60% étaient des hommes, l'âge moyen était de 39,1 (± 12,8) ans et 50% des sujets étudiés ont été testés positifs pour H. pylori COMPARARISON AVEC ET les deux technologies singleplex (RT-PCR) et multiplex (plate-forme solide) ont montré une forte expression (expression de plus de 1000 se lit comme suit dans le DGE et 7 fois au-dessus du contrôle endogène dans la RT-PCR) des 15 miARN (figures 4 et 5) Les mêmes échantillons d'ARN ont été analysés par deux plates-formes différentes: DGE et RT-PCR - Life Technologies.. Les résultats des expériences ont été comparées et ont été observées régression linéaire entre 2 -ΔCt et racine carrée du nombre de comptage lu. corrélation de Pearson est élevé à 83,9% avec un test statistique significative (P < 0,05). et validé les résultats SOLIDES En outre, les résultats DGE ont été comparés à la quantification de la gamme d'expression des miARN dans 10 sujets sains, par Temps réel PCR. La régression de la moyenne des tests de RT-PCR par rapport Impossible d'observer miARN avec une grande variation interindividuelle, pour exempla miR-21, et un autre avec une faible variation interindividuelle, par exemple motif d'expression légèrement variable (miR-29b, miR-29c, miR-19b, miR-31, miR-148a, miR-451). Discussion miARN régulent la majorité des gènes humains; cependant, seuls quelques miARN ont eu leurs objectifs et fonctions spécifiques identifiés [14]. Dans notre étude, l'échantillon de l'estomac a été obtenu à partir d'un seul individu sans néoplasie de l'estomac ou d'autres conditions de pré-néoplasiques tels que l'atrophie, la métaplasie ou une dysplasie. lésions pré-cancéreuses telles que la gastrite conduisent à génomique hypo-méthylation dans l'estomac qui pourrait modifier le profil d'expression des miARN [15]. L'échantillon a été obtenu à partir du tissu normal d'un patient, sans aucune pathologie, ce qui a permis d'éviter le risque de prélèvement d'un échantillon de tissu apparemment normale avec des micro-invasion des cellules tumorigènes stade précoce comme on pouvait survenir chez des patients avec l'une des pathologies ci-dessus. Cette étude est le premier séquençage ultra-haut débit de miARN dans l'estomac humain physiologique normal. Seulement 5,06% des miARN identifiés dans le tissu gastrique avait déjà été détectée dans d'autres tissus et catalogués dans des banques de données bioinformatiques tels que microRNA.org [10]. Nous nous attendons à ce groupe de miARN d'être des régulateurs de gènes de ménage, qui sont abondants dans les tissus humains. Un autre 7,84% des miARN avait aucune correspondance dans les bases de données d'expression de miARN et pourrait représenter miARN qui sont spécifiques au système digestif ou de l'estomac. Des études similaires ont été réalisées avec d'autres tissus normaux, tels que la bouche, du pharynx, oesophage, de l'anus et de l'intestin. Nous avons comparé ces données avec les données d'expression de miARN pour définir le motif d'expression du tissu de l'estomac. Nous avons trouvé des niveaux élevés d'expression dans 15 miARN, dont 13 avaient déjà été identifiées comme hautement exprimé dans d'autres tissus. L'expression de mir-148a et mir-192 avait été identifié dans d'autres tissus humains normaux et cancéreux, mais n'a pas été surexprimé. MiR-192 avait déjà été détectée dans les tissus gastro-intestinaux tels que le côlon, l'iléon, le duodénum, l'intestin grêle, de l'estomac, le pancréas et le foie [16]. expression basale de miR-148a a été observée dans le tissu conjonctif et du tissu endocrinien [17]. Récemment, mir-148a a été trouvé pour être réprimée dans ombilicaux cellules de sang de cordon [18] et réduits au silence par hyperméthylation dans les tumeurs du côlon [19]. Nous avons observé une forte expression de la grappe mir-200 (a et b) dans le cardia gastrique observée dans les îlots de Langerhans [11]. Dans une expérience de microréseau, mir-200a et miR-200b ont été détectés à des niveaux faibles dans les tissus gastro-intestinaux, mais à des niveaux élevés dans le côlon, de l'estomac et du pancréas [16]. A microARN publiées récemment atlas d'expression ont montré que ce miARN est caractéristique pour le tissu endocrinien [17]. Des résultats récents montrent que la faible expression du cluster mir-200 est en corrélation avec le cancer de l'ovaire [19], [20]. Par conséquent, le cluster mir-200 peut être important dans le maintien de l'intégrité des tissus digestifs tels que cardia gastrique parce que cette forte expression régulée à la hausse l'expression de e-cadhérine, la protéine responsable de l'organisation de l'architecture du tissu épithélial. De plus, les résultats suggèrent fortement un rôle important de la famille miR-200 dans la répression de l'épithélium-mésenchyme transition (EMT) et la progression du cancer [21]. Tableau 1 et 2 montrent le nombre de cibles possibles pour chaque maturité miARN fortement exprimé. Le tableau 2 montre certaines cibles de miARN prévues par le TargetScan contre les familles de miARN conservées. A l'exception de miR-451, tous partagé au moins deux autres gènes cibles. Les gènes ANKD52 De nombreux miARN peuvent réguler la traduction des protéines qui agissent dans la prolifération des tissus et la structuration des tissus tels que mir-200a (qui peut cibler intégrine) et mir-145 (qui peut interagir avec ERBB4 Plusieurs observations lien miARN au cancer. Premièrement, de nombreux miARN sont impliqués dans la prolifération cellulaire et l'apoptose. Deuxièmement, de nombreux loci miARN sont situés dans des sites fragiles du génome humain, les régions qui sont souvent amplifiés ou supprimés dans les néoplasies humaines et provoquent de grandes différences dans l'expression des miARN par rapport aux tissus normaux [21], [22], [23], [24 ]. la technique PCR en temps réel (qui utilise la quantification relative) a confirmé 15 miARN identifiés comme présentant la plus haute expression de la plate-forme solide (qui est basé sur des chiffres absolus) (figures 4 et 5). Par conséquent, ces miARN peuvent être considérés comme étant surexprimée [25], [26], [27], [28]. La corrélation entre la DGE et les dosages RT-PCR était claire et statistique significative. Et l'expérience DGE pourrait être considéré comme représentatif des tissus échantillons gastriques isolés à partir de 10 sujets de santé et définissent une partie du motif d'expression du tissu gastrique sain. Les résultats des deux méthodes indiquent que miRNA-21 était le plus fortement exprimés dans les tissus du cardia gastrique. Cette miARN est également distribué dans d'autres tissus humains (par exemple, les cellules dendritiques, les cellules T, le pancréas) et pourrait être impliqué dans la régulation de l'expression des gènes de ménage ( ATPAF1 La plate-forme SOLiD ont montré que miR-192 et 148a sont spécifiques au tissu gastrique. En outre, PCR en temps réel a confirmé que ces miARN sont sur-exprimés. Ainsi, dans l'ensemble, ces miARN sont susceptibles de réguler l'expression de gènes liés à l'homéostasie tissulaire gastrique. Par conséquent, les faibles niveaux d'expression de ces miARN pourraient être liées à l'élaboration d'une néoplasie de l'estomac. L'utilisation potentielle de miARN-192 et miRNA-148a comme marqueurs de risque dans le cancer gastrique pourraient être mieux étudiés à travers l'analyse des miRnomes de différents types histologiques de cancer de l'estomac. Comprendre les processus réglementaires qui agissent dans l'humain l'estomac sera important dans la lutte contre le cancer de l'estomac, qui est la deuxième cause de mortalité par cancer dans le monde entier. Matériaux et méthodes MATÉRIEL BIOLOGIQUE le cardia est une zone microscopique qui se trouve normalement dans la partie la plus proximale de l'estomac, à proximité de l'ouverture de l'œsophage (orifice cardiaque ou cardia) et contient les glandes cardiaques. Notre échantillon de tissus frais pour le séquençage ultra profond a été obtenu à partir d'une biopsie gastroscopique (~ 4 mm 3). Le patient était âgé de 33 ans, sans signe de cancer et d'une jonction gastro-oesophagienne normale. L'observation macroscopique du tissu n'a montré aucun signe de lésions, et l'examen histologique a confirmé les conditions normales et en bonne santé. Pour confirmer les résultats du séquençage ultra-profond, des échantillons de cardia, l'orifice près cardiaque, de 10 plus, les personnes en bonne santé ont été également obtenus par biopsies endoscopiques (~ 4 mm 3) et, après examen histologique excluant les anomalies, ont été analysés par PCR en temps réel. H. pylori de l'infection a été diagnostiquée sur la base de l'aspect typique de la bactérie sur la couche de mucus recouvrant la muqueuse gastrique biopsies antrales ont été prélevés pour une évaluation histologique en raison de la densité plus élevée des bactéries dans l'antre gastrique et à l'excellente sensibilité de cette méthode de diagnostic , selon les critères internationaux établis pour leur identification [12], [13] (des détails supplémentaires sont fournis dans le tableau S2). ÉTHIQUE DÉCLARATION Le consentement éclairé écrit a été obtenu de tous les patients, et l'étude a été approuvée par le Comité de Ética em Pesquisa (CEP) de l'hôpital Universitário João Barros Barreto (HUJBB) -. Université fédérale de Pará (UFPA) (numéro de protocole 14052004 /HUJBB) miRNA BIBLIOTHÈQUE le petit ARN total a été obtenu à partir de l'échantillon de tissu en utilisant le kit d'isolation MIRVANA (Ambion Inc., Etats-Unis). La concentration et la qualité ont été déterminées à l'aide d'un spectrophotomètre Nanodrop 1000, et la purification et la sélection de la taille ont été effectuées en utilisant une électrophorèse sur gel de Polyacrylamide à 6%. En utilisant le solide petit kit d'expression de l'ARN (Ambion Inc., Etats-Unis), 200 ng d'ARN de petite taille 150-200 pb ont été utilisés comme matrice pour obtenir la bibliothèque miARN. Tous les miARN de la bibliothèque ont été marqués avec des amorces d'amplification uniques et spécifiques, connus sous le système de code à barres (Life Technologies, CA, États-Unis). Ensuite, 50 pg de la bibliothèque a été mis en commun avec sept autres bibliothèques miARN à la même concentration. Une fraction du pool de la bibliothèque (0,1 pg) a été amplifié et fixé sur des billes magnétiques en utilisant une émulsion par PCR. Le produit ePCR a été déposé sur une seule lame et soumis à la réaction de séquençage SOLiD multiplex. La (version 2.0) système de séquençage SOLiD (Life Technologies) a été utilisé pour générer lit qui était de 35 pb. La seconde étape consistait à décoder le code à barres correspondant à chaque séquence de perle avec l'identité de l'échantillon. Toutes les petites séquences d'ARN du cardia sont disponibles dans le NCBI lire les séquences d'archives (SRA012099). L'analyse de séquence a été réalisée en utilisant le système SOLiD Outil petit ARN Analyse (Life Technologies) et MiRanalyzer [8]. Tout d'abord, nous avons filtré sur toutes les séquences qui correspondent à des contaminants d'ARN tels que ARNt, ARNr, répétitions d'ADN et des molécules d'adaptateur. Après l'exclusion de contaminant lit, nous avons aligné toutes les séquences contre miRNA séquences précurseurs (miRBase ver. 12) et seulement inclus les lectures que appariés séquences matures miRNA [9]. Pour comparer ces données d'expression avec ceux d'autres tissus humains, des données d'expression de miARN ont été importées de la base de données microRNA.org, et l'expression de chaque maturité miRNA a été normalisé par le nombre de lecture totale [10]. L'analyse graphique a été réalisée en utilisant Genepattern 10. Les relations de traitement miRNA biologique ont été prédits en utilisant miRNApath (http://lgmb.fmrp.usp.br/mirnapath/tools.php). Biopsy échantillons de tissus ont été prélevés cardia chez 10 patients sains. Après la collecte, les échantillons ont été immédiatement traités et stockés à -80 ° C jusqu'à l'extraction d'ARN. L'ARN total a été extrait par homogénéisation de 40 milligrammes de tissu congelé, suivie par l'isolement de l'ARN par le réactif Trizol (Life Technologies) selon les instructions du fabricant. La concentration et la qualité de miARN ont été déterminées à l'aide d'un spectrophotomètre Nanodrop 1000 (ND-1000; Nanodrop Technologies, Wilmington, DE). L'ARN total a été transcrit inverse en utilisant un TaqMan @ kit microARN transcription inverse (Life Technologies). L'analyse des niveaux miARN a été effectuée sur un système de PCR en temps réel 7500 (Life Technologies) avec des dosages TaqMan miARN selon le les instructions du fabricant (Life Technologies) en utilisant des amorces conçues avec Primer express (Life Technologies). Le niveau de trois contrôles endogènes humains (Z30, RNU19 et RNU6B - calibrateurs) signifie expression. A été utilisé comme un contrôle interne dans toutes les expériences miRNA pour permettre la comparaison des résultats d'expression Les niveaux d'expression élevés de miARN identifiés par séquençage ultra-profond (par ordre décroissant: miR-29c, miR-21, miR-148a, miR-29a, miR-24, miR-29b, miR-192, miR-451, miR-145, miR-31, miR-200a, miR-19b, miR-200b, laissez-7b et miR-26a) ont été validés avec les dosages TaqMan miARN (Life Technologies). Ces analyses ont été utilisés pour mesurer les niveaux d'expression des miARN matures et la variation inter-individuelle dans les 10 échantillons de tissu cardia sain. Les données d'expression de chaque miARN matures ont été normalisées au niveau d'expression moyenne des trois contrôles endogènes humains (Z30, RNU19 et RNU6B). Les niveaux relatifs d'expression de miARN ont ensuite été calculés par la méthode du cycle de comparaison de seuil (Ct) (2 -ΔCt). test de corrélation a été réalisée en utilisant la méthode Pearson (SPSS v.12). Informations complémentaires Remerciements Le auteurs remercient D. Calcagno et S. Demaschki pour leurs commentaires, idées et aide.
2009 a démontré que la majorité des gènes humains sont sous le contrôle des miARN. Le > 45.000 miRNA sites cibles au sein de 3'UTRs humains sont conservés, et > 60% des gènes codant pour des protéines humaines ont été sous pression sélective pour maintenir appariements à miARN [1]. miARN sont des séquences de petite taille non codants de 17 à 25 pb qui régulent l'expression des gènes en se liant à l'extrémité 3 'des ARNm cibles, ce qui entraîne l'inhibition de la traduction de l'ARNm [2], [3]. Environ 14 000 miARN ont été identifiés dans les animaux, les plantes et les champignons [4], [5], [6]
Résultats
ULTRA-DEEP SEQUENCING
PCR
, selon les critères internationaux établis pour leur identification [12], [13] (tableau 2) (détails supplémentaires sont fournis dans le tableau S2).
DGE RT-PCR
racine carrée du nombre de comptage lu. corrélation de Pearson est élevé à 68,4% avec un test statistique significative (P < 0,05).
et UBN2
, sont des cibles de dix des quatorze miARN analysés, tandis que le gène TNRC6B
est de neuf miARN, et les gènes EPS15
, NFAT5
, BACH2
, BRWD1
, NUFIP2
, PTEN
, CDK6
et PTPRD DD6
, sont des cibles de huit miARN. Ce résultat suggère que ces miARN sont des candidats forts pour être réduits au silence dans la région du cardia. La validation expérimentale de ces gènes, suivie d'une analyse de la fonction de chacun peut révéler le rôle physiologique de ces miARN dans le tissu gastrique normale.
ARNm). Plusieurs cibles d'ARNm prévus se sont révélés être communs à plusieurs miARN fortement exprimés. Par exemple, le (récepteur 5-hydroxytryptamine [sérotonine]) HTR4
et AFF2
ARNm ( La famille de AF4 /FMR2, membre 2) ont été prévus pour être des cibles de 13 des 15 miARN les plus fortement exprimés. Cinq autres ARNm, y compris IGF-1
(facteur de croissance insulinomimétique de type 1 [somatomédine C]), étaient des cibles prédites ordinaires de 12 miARN. Huit miARN avaient 222 cibles prédites en commun; par exemple, mir-29b a été prédit pour cibler 196 de ceux-ci. Cinq miARN (19b, 29a, 29b, 29c et 148a) ont partagé 70 prédit cibles, dont certains ( CDK6
, PTEN
, IGF1
, FRS2
, PDGFRA
, PIK3R1
et MXD1
) réguler la prolifération cellulaire et la suppression tumorale.
, KIF3A
, CYBRD1
).
SOLiD SEQUENCING ULTRA-PROFONDE ET ANALYSE DES DONNÉES
miRNA REAL TIME PCR (VALIDATION)
Tableau S1.
identité et des données d'abondance pour tous les miARN connus dans le dataset de séquence SOLiD dans l'estomac humain et la base de données miRamda
doi:. 10.1371 /journal.pone.0013205.s001
(0.44 MB PDF)
Tableau S2.
La description et les résultats de RT-PCR de 10 échantillons provenant d'individus sains obtenus par biopsies endoscopiques. Les échantillons de taille est d'environ ~4 mm3. Pour tous les échantillons ont été effectués un examen histologique et la détection de H. pylori
doi:. 10.1371 /journal.pone.0013205.s002
(0.03 MB XLS)
 Les microbes pourraient prédire des issues fatales chez les patients ventilés COVID-19
Les microbes pourraient prédire des issues fatales chez les patients ventilés COVID-19
 Un modèle de souris nouveau-né donne des indices sur la cause d'une maladie intestinale dévastatrice chez les prématurés anémiques
Un modèle de souris nouveau-né donne des indices sur la cause d'une maladie intestinale dévastatrice chez les prématurés anémiques
 Ce que vous mangez peut changer la façon dont les antibiotiques affectent votre intestin
Ce que vous mangez peut changer la façon dont les antibiotiques affectent votre intestin
 Les microbes intestinaux pourraient être liés à la dépression
Les microbes intestinaux pourraient être liés à la dépression
 Comment booster votre système immunitaire pour lutter contre le coronavirus
Comment booster votre système immunitaire pour lutter contre le coronavirus
 Le microbiome intestinal est également une réalité dans la vie fœtale
Le microbiome intestinal est également une réalité dans la vie fœtale
Une étude publiée dans le Journal dinvestigation clinique montre que chez les souris et les humains, lintestin fœtal a son propre microbiome, qui est probablement dérivé directement de lorganisme ma
Le microbiome intestinal est également une réalité dans la vie fœtale
Le microbiome intestinal est également une réalité dans la vie fœtale
Une étude publiée dans le Journal dinvestigation clinique montre que chez les souris et les humains, lintestin fœtal a son propre microbiome, qui est probablement dérivé directement de lorganisme ma
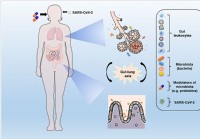 La modulation du microbiote et le rétablissement de l'eubiose pourraient aider à freiner les complications du COVID-19
La maladie à coronavirus (COVID-19), causée par le coronavirus 2 du syndrome respiratoire aigu sévère (SARS-CoV-2), affecte de nombreux organes du corps. En plus dêtre une maladie respiratoire, il peu
La modulation du microbiote et le rétablissement de l'eubiose pourraient aider à freiner les complications du COVID-19
La maladie à coronavirus (COVID-19), causée par le coronavirus 2 du syndrome respiratoire aigu sévère (SARS-CoV-2), affecte de nombreux organes du corps. En plus dêtre une maladie respiratoire, il peu
 Selon une étude, les cellules immunitaires intestinales pourraient être responsables des changements métaboliques
Une nouvelle étude a montré que les cellules immunitaires dans lintestin pourraient être liées au taux de métabolisme. Les résultats de la nouvelle étude intitulée, « Les cellules T intraépithéliales
Selon une étude, les cellules immunitaires intestinales pourraient être responsables des changements métaboliques
Une nouvelle étude a montré que les cellules immunitaires dans lintestin pourraient être liées au taux de métabolisme. Les résultats de la nouvelle étude intitulée, « Les cellules T intraépithéliales