FDA on tutkinut NMDA:ta ja muita nitrosamiiniepäpuhtauksia jo jonkin aikaa erityisesti verenpainelääkkeissä, joita kutsutaan angiotensiini II -reseptorisalpaajiksi. Korkeat nitrosamiinipitoisuudet ovat johtaneet useiden näiden lääkkeiden merkkien vetämiseen markkinoilta. Tällä hetkellä ranitidiinimerkkeissä havaitut nitrosamiinipitoisuudet ovat alhaiset, ja FDA julkaisee lausuntoja palautuksesta. Tämä oli varoitusraportti.
Raportti sanoo, ”Potilaiden pitäisi voida luottaa siihen, että heidän lääkkeensä ovat mahdollisimman turvallisia ja että niiden ottamisesta saatavat hyödyt ovat suuremmat kuin terveydelle aiheutuvat riskit. Vaikka NDMA voi vahingoittaa suuria määriä, FDA:n ranitidiinipitoisuudet alustavien testien perusteella ylittävät tuskin määrät, joita saatat odottaa tavallisista elintarvikkeista. ”
Ranitidiini on laajalti käytetty reseptilääke ja reseptilääke, joka kuuluu H2 (histamiini-2) -estoaineryhmään ja on hyödyllinen mahahapon erityksen vähentämisessä. Sitä käytetään närästyksen hoitoon, hapon nieleminen jne. maha- ja pohjukaissuolihaavan ennaltaehkäisy ja GERD:n (gastroesofageaalinen refluksitauti) hoito ja hoito.
Toistaiseksi FDA tutkii NDMA:n lähdettä ranitidiinin valmistuksessa ja toivoo, että jos se poistetaan, lääke voidaan julistaa jälleen turvalliseksi. Raportti sanoo, ”FDA ryhtyy tarvittaviin toimenpiteisiin meneillään olevan tutkimuksen tulosten perusteella. Virasto antaa lisätietoja, kun ne tulevat saataville. ”
Varoitus toistaa, että yksilöt eivät lopeta ranitidiinin käyttöä tämän varoituksen perusteella. Henkilöiden, jotka haluavat lopettaa ranitidiinilääkityksen, on keskusteltava terveydenhuollon tarjoajien kanssa vaihtoehtoisista hoitovaihtoehdoista, sanoo FDA.
FDA lisäsi varoitukseen, "Kuluttajien ja terveydenhuollon ammattilaisten tulee ilmoittaa kaikista ranitidiinin aiheuttamista haittavaikutuksista FDA:n MedWatch -ohjelmaan auttaakseen virastoa ymmärtämään paremmin ongelman laajuuden." He pyytävät kuluttajia täyttämään ja toimittamaan raportin verkossa osoitteessa www.fda.gov/medwatch/report.htm ja täyttämään asianmukaisen lomakkeen ja lähettämään faksin numeroon 1-800-FDA-0178.
24. syyskuuta, FDA julkaisi jälleen varoituksen, joka varoitti terveydenhuollon ammattilaisia ja potilaita, että he olivat muistuttaneet 14 erää ranitidiinikapseleita, jotka Sandoz Inc. jakoi. Tämä muistutus johtui erissä olevista NDMA -määristä.
FDA:n komissaari Ned Sharpless, M.D., lausunnossaan sanoi, ”FDA on sitoutunut varmistamaan, että amerikkalaisten ottamat lääkkeet ovat turvallisia ja tehokkaita. Aloitimme ranitidiinituotteiden testaamisen heti sen jälkeen, kun saimme tietää mahdollisesta epäpuhtaudesta. Kun havaitsemme potilaiden mahdollisia riskejä aiheuttavien lääkkeiden laadun heikkenemisen, FDA tekee kaikkensa ymmärtääkseen asian ja antaakseen parhaan suosituksen yleisölle mahdollisimman nopeasti ja tarkasti. ” "Jatkamme tutkimuksia ja pyrimme varmistamaan, että tämäntyyppiset epäpuhtaudet eivät ylitä sallittuja rajoja, jotta potilaat voivat jatkaa tarvitsemiensa lääkkeiden ottamista huoletta, " hän lisäsi.
Toisessa raportissa FDA toimitti potilaille ja terveydenhuollon ammattilaisille tietoja Sandozin tuottamasta ranitidiinista sanomalla, että jos potilas käytti jotakin muistutetuista lääkkeistä, hänen tulee noudattaa FDA:n verkkosivustolla olevia palautusohjeita. Raportissa lisättiin, että ne potilaat, jotka eivät ole ranitidiinimerkkejä, joita ei palauteta, voivat jatkaa sitä. Raportti lisäsi, "On tärkeää muistaa, että kaikkea Yhdysvalloissa markkinoitavaa ranitidiinia ei palauteta." Raportin mukaan potilaat, jotka käyttävät OTC -ranitidiinia, voivat harkita muita vaihtoehtoja oireilleen.
Janet Woodcock, M.D., FDA:n lääkkeiden arviointi- ja tutkimuskeskuksen johtaja, lausunnossaan sanoi, "Jatkamme tutkimustamme yhdessä kansainvälisten kollegoidemme kanssa, ja pidämme amerikkalaista yleisöä ajan tasalla mahdollisista lisäkutsuista sekä ranitidiinivalmisteiden ottamisesta mahdollisesti aiheutuvista riskeistä. ”
NDMA -tasojen havaitsemiseksi FDA on lähettänyt sääntelyviranomaisille ja valmistajille protokollan. Näiden valmistajien tekemät ranitidiinin NDMA -pitoisuudet on ilmoitettava näiden testien avulla. Lisäksi heidän on lähetettävä näytteet FDA:lle testattavaksi viraston tiedemiehille.
 Suoliston mikrobiomi on todellisuutta myös sikiön elämässä
Suoliston mikrobiomi on todellisuutta myös sikiön elämässä
 Ikkasairaus ja ruokatorven ja mahasyövän riski
Ikkasairaus ja ruokatorven ja mahasyövän riski
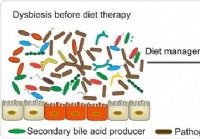 Suoliston mikrobiomi ja IBD - yhteys ehkä ruokavaliossa, sanoo tutkimus
Suoliston mikrobiomi ja IBD - yhteys ehkä ruokavaliossa, sanoo tutkimus
 Kasvipohjainen ruokavalio voi parantaa nivelreuman
Kasvipohjainen ruokavalio voi parantaa nivelreuman
 Tutkijat löytävät uuden tavan suojautua sairauksilta MS -mallissa
Tutkijat löytävät uuden tavan suojautua sairauksilta MS -mallissa
 Keliakian hoito
Keliakian hoito
 Valkosolut ja niiden rooli aivoissa
Uraauurtavassa tutkimuksessa ryhmä kansainvälisiä tutkijoita on havainnut, että aivoissa on erityisiä sisäisiä immuunisoluja, jotka auttavat normaalia aivojen kehitystä ja joilla on rooli tietyissä ne
Valkosolut ja niiden rooli aivoissa
Uraauurtavassa tutkimuksessa ryhmä kansainvälisiä tutkijoita on havainnut, että aivoissa on erityisiä sisäisiä immuunisoluja, jotka auttavat normaalia aivojen kehitystä ja joilla on rooli tietyissä ne
 Uusi strategia voi vahvistaa suoliston ja aivojen välistä viestintää
Suoliston ja aivojen välinen viestintäjärjestelmä tunnetaan suolen ja aivojen akselina ja se on vakiintunut. Nyt, tutkijat ovat kehittäneet strategian, joka lisää suolen ja kehon välisen viestinnän mä
Uusi strategia voi vahvistaa suoliston ja aivojen välistä viestintää
Suoliston ja aivojen välinen viestintäjärjestelmä tunnetaan suolen ja aivojen akselina ja se on vakiintunut. Nyt, tutkijat ovat kehittäneet strategian, joka lisää suolen ja kehon välisen viestinnän mä
 Punkit kantavat nyt useita sairauksia,
sanoo uusi tutkimus Uusi tutkimus julkaistiin lehdessä mBio raportoi, että punkit kantavat paljon enemmän tauteja aiheuttavia aineita kuin vain ne, jotka ovat vastuussa Lymen taudin aiheuttamisesta.
Punkit kantavat nyt useita sairauksia,
sanoo uusi tutkimus Uusi tutkimus julkaistiin lehdessä mBio raportoi, että punkit kantavat paljon enemmän tauteja aiheuttavia aineita kuin vain ne, jotka ovat vastuussa Lymen taudin aiheuttamisesta.