 Personer med ubehandlet Wilsons sygdom kan have en forventet levetid på 40 år; tidlig diagnose og behandling kan dog forlænge levetiden.
Personer med ubehandlet Wilsons sygdom kan have en forventet levetid på 40 år; tidlig diagnose og behandling kan dog forlænge levetiden. Wilsons sygdom er en meget sjælden genetisk lidelse nedarvet i et autosomalt recessivt mønster, som kan overføres til næste generation fra forældre, der bærer en eller begge kopier af det berørte gen. Det anslås, at 1 ud af 40.000 mennesker på verdensplan er ramt af Wilsons sygdom.
For at udvikle Wilsons sygdom skal du arve to kopier af det defekte gen, en fra hver forælder. Hvis du kun arver én kopi af det defekte gen, så vil du være bærer og give genet videre til næste generation, men du vil ikke udvikle sygdommen.
Normalt udvikler symptomer på Wilsons sygdom mellem 12 og 23 års alderen, og ubehandlede mennesker kan have en forventet levetid på 40 år . Men tidlig diagnose, efterfulgt af korrekt behandling, kan forlænge levetiden.
Wilsons sygdom er forårsaget af en defekt i ATP7B gen, som forhindrer metabolisering og eliminering af kobber fra kroppen, hvilket potentielt udvikler sig til en livstruende tilstand.
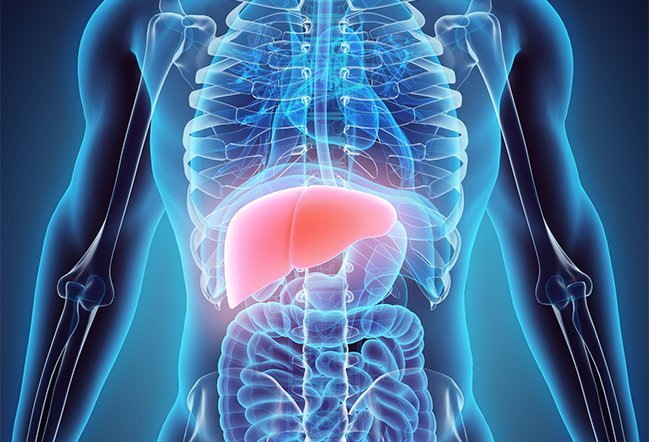
Selvom din krop har brug for en lille mængde kobber til visse kropsfunktioner, er en overskydende mængde af dette mineral giftig og kan føre til komplikationer. Leveren spiller en stor rolle i metabolismen af kobber med overskydende kobber overført til galden, som senere elimineres gennem afføring.
Wilsons sygdom påvirker forskellige organer, hvilket resulterer i forskellige symptomer, der kan omfatte:
Diagnosen af Wilsons sygdom udføres ikke let i de indledende stadier, fordi de forårsagede symptomer er korreleret med mange andre medicinske tilstande, især med tilstande, der involverer lever og nerver.
Læger diagnosticerer Wilsons sygdom ved følgende:
Læger udfører magnetisk resonansbilleddannelse og computertomografi for at opdage eventuelle hjerneabnormiteter. Selvom disse fund ikke hjælper med den indledende diagnose af sygdommen, vil de hjælpe med at bekræfte den og bestemme det aktuelle stadium.
Læger kan udføre en leverbiopsi for at bestemme omfanget af kobberaflejring og mulig leverskade.
Læger udfører en procedure, hvor de trækker et lille stykke af levervævet ud ved at bruge en nål. Denne procedure udføres under påvirkning af anæstesi. Læger studerer det ekstraherede levervæv under et mikroskop for at tjekke for kobberaflejringer.
Wilsons sygdom kan ikke forebygges, fordi den føres gennem gener fra forældre uden kendt helbredelse. Du kan dog bremse udviklingen af sygdommen ved tidlig diagnose og behandling af symptomerne.
Behandlingsmuligheder for Wilsons sygdom omfatter:
 Cøliaki 101 – En begyndervejledning til helbredelse
Cøliaki har mange uudtalte forfærdelige sandheder... En af dem er denne:Det tager i gennemsnit 4 år at få en Cøliaki-diagnose, og forskningen viser ofte, at over 2 år senere er de fleste patienter
Cøliaki 101 – En begyndervejledning til helbredelse
Cøliaki har mange uudtalte forfærdelige sandheder... En af dem er denne:Det tager i gennemsnit 4 år at få en Cøliaki-diagnose, og forskningen viser ofte, at over 2 år senere er de fleste patienter
 Typer af ernæringsrør og deres anvendelse
En plastikslange er et medicinsk udstyr, der bruges til at fodre en person, der ikke er i stand til at tage mad eller drikke sikkert gennem munden. Dette problem kan skyldes synkebesvær, en ændret men
Typer af ernæringsrør og deres anvendelse
En plastikslange er et medicinsk udstyr, der bruges til at fodre en person, der ikke er i stand til at tage mad eller drikke sikkert gennem munden. Dette problem kan skyldes synkebesvær, en ændret men
 Lad os blive snavsede med SIBO
Jeg har udskudt at blogge om dette. Hovedsageligt fordi jeg ikke tror på, at en one-size fits all-tilgang virker. Jeg tror ikke, forskningen er der, hvor vi har brug for den, for at jeg kan give dig d
Lad os blive snavsede med SIBO
Jeg har udskudt at blogge om dette. Hovedsageligt fordi jeg ikke tror på, at en one-size fits all-tilgang virker. Jeg tror ikke, forskningen er der, hvor vi har brug for den, for at jeg kan give dig d