Verordnung des Aktin-Zytoskelett in Helicobacter pylori
-induzierte Migration und invasive Wachstum von Magenepithelzellen
Zusammenfassung
Dynamische Umlagerung des Aktin-Zytoskelett ist ein bedeutendes Merkmal von Helicobacter pylori
(H. pylori
) infiziert. In Anbetracht der zellulären Mechanismen, die Typ IV-Sekretionssystem (T4SS) und die Effektor-Protein-Cytotoxin-assoziierte Gen A (CagA) von H. pylori
sind gut untersucht Initiatoren verschiedener Signaltransduktionswege in Wirtszellen-Targeting-Kinasen, Adapterproteine , GTPasen, Actin-Bindung und andere in der Regulation des Gitters Aktin beteiligten Proteine. In diesem Bericht fassen wir jüngsten Erkenntnisse, wie
H. pylori interagiert funktionell mit dem komplexen Signalnetzwerk, das die Aktin-Zytoskelett motiler und invasive Magenepithelzellen steuert.
Schlüsselwörter Helicobacter pylori
Typ IV Sekretionssystem CagA Aktinzytoskelett Bewertung
die kontinuierliche Reorganisation und den Umsatz des Aktin-Zytoskelett ist ein fundamentaler Prozess in der Regulation der Zelladhäsion an Nachbarzellen und der extrazellulären Matrix (ECM), Phagozytose, Zellform oder Migration. Im Allgemeinen existiert Aktin in Zellen als monomere globular Actin (G-Actin) und filamentöses Aktin (F-Aktin), die bei der Polymerisation von G-Actin-Monomere in einem definierten Direktionalität gebildet werden. Eine Vielzahl von Upstream-Signalmoleküle, einschließlich der Zelladhäsionsmolekül E-Cadherin, Integrine, Komponenten der ECM oder Stimuli, wie Tumor-Nekrose-Faktor alpha (TNF-α) und Lysophosphatidsäure (LPA) sind bei der Übertragung von extrazellulären Signalen bekannt an das Aktin-Zytoskelett schnelle Reaktion auf eine sich verändernde Umwelt (Abbildung 1A) ermöglicht. Daher Umbau des Aktin-Zytoskelett-Architektur auf einer großen Gruppe ist abhängig von Signalmolekülen, die die Montage des Aktin-Netzwerk an Aktin und modulieren binden (siehe [1] für eine umfassende Übersicht). Figur 1 Signalübertragungswege in der Regulation des Actin-Zytoskeletts beteiligt. (A) Die Bildung von Aktin-abhängige Strukturen, wie Stressfasern, fokale Adhäsionen, Lamellipodien und Filopodien wird durch Zelloberflächenmoleküle im Bereich von E-Cadherin und Integrine an Rezeptoren für kleine Bauteile gesteuert (zB
. TNF-α oder LPA) ermöglicht die Übertragung von extrazellulären Stimuli auf das Aktin-Zytoskelett. Der Rho-GTPasen RhoA, Rac1 und Cdc42 sind Schlüsselelemente in der Regulation der Aktin-Filamente. Rac1 und Cdc42 induzieren Aktin-Polymerisation durch WASP /WAVE Familienmitglieder und WIPs die Arp2 /3-Komplex stimulieren. RhoA regelt DIA1 /Profilin und die ROCK /MLC Wege Polymerisation von F-Actin zu fördern. (B) Fokaladhäsionen sind wichtige Strukturen in die Verknüpfung der ECM an die intrazelluläre Aktin-Zytoskelett über α und β-Integrin-Heterodimeren. Der extrazelluläre Teil der Integrine bindet an Proteine der ECM, während die intrazelluläre Domäne eine Vielzahl von intrazellulären Signal (FAK, Src, etc
.) Und Adapterproteine (Talin, paxillin, Vinculin oder p130CAS usw
.), um das Aktin-Zytoskelett zu verbinden.
Unter Aktin-abhängige zelluläre Prozesse, erfordert effiziente Zellmigration eine koordinierte Umlagerung des Aktin-Gitter in motile Zellen. Polymerisation von F-Aktin an Zell Vorsprüngen löst die Bildung von folienähnlichen Lamellipodien und stangenartigen Filopodien drängen migrierenden Zellen [2, 3]. Zusätzlich Bildung von kontraktilen Strukturen durch Wechselwirkung mit Myosin II von Aktin zieht den Zellenkörper über den ECM. Diese Verfahren umfassen ein breites Spektrum an Aktin bindenden Proteinen (z.B. Cortactin, α-Actinin, Fascin, Profilin, Filamin, etc
.), Die Actinstabilisierung beitragen, Bündelung und Verzweigung, ein komplexes Netzwerk bilden. Signalwege Aktin Umlagerung Modulation sind komplex und haben in mehreren ausgezeichneten Bewertungen bedeckt worden [4-6]. Fasst man die wichtigsten Erkenntnisse in einem simplen Modell (Abbildung 1A), Signalwege an Zelloberflächenrezeptoren initiiert zu fördern deutliche Membranausstülpungen konvergieren auf Rho GTPasen als die wichtigsten Elemente der Signaltransduktion. Eines der am besten charakterisierte Rho-GTPase Familienmitglieder ist RhoA die Bildung von Stressfasern und focal adhesion Montage regulieren, während Rac und Cdc42 hauptsächlich beteiligt sind in der Membran Kräuseln und die Bildung von Filopodien bzw. [4]. Rac1 und Cdc42 kann Aktin-Polymerisation durch Mitglieder des Wiskott-Aldrich-Syndrom-Protein (WASP) Familie und WASP-interagierende Proteine (WIPs) induzieren. Die WASP Familie der Aktin-Nukleation fördernden Faktoren (NPFs) umfasst WASP, N-WASP und vier Formen von WASP verprolin homologe Protein (WAVE). Durch eine konservierte C-terminale Domäne, stimulieren WASP Proteine die Aktin-verwandte Proteine 2/3 (Arp2 /3) -Komplex Aktivität Aktinfilamente zu nukleieren und an ihren freien Enden mit Widerhaken zu verlängern. Stress Faseranordnung und Zusammenziehung werden überwiegend durch RhoA induziert [7], wie oben erwähnt, die DIA1 /Profilin steuert Polymerisation von F-Aktin zu fördern [8]. Ein weiterer Mechanismus beinhaltet Rho-induzierte Rho-assoziierte Serin /Threonin-Kinase (ROCK) als wichtiger Downstream-Effektor-Myosin leichte Kette (MLC) Phosphorylierung [9] (1A) zu induzieren.
Üblicherweise kontraktilen Stressfasern zum Plasma befestigen Membran an naszierenden fokalen Adhäsionen, die durch α und β-Integrin heterodimeren Rezeptoren (1B) stabilisiert sind. Überbrückung der ECM an das Aktin-Zytoskelett bindet das Integrin ectodomain direkt an ECM-Proteine (zB Fibronektin), während die intrazelluläre Domäne an das Aktin-Zytoskelett über rekrutiert Adapter und Signalproteinen einschließlich focal adhesion kinase (FAK) verbunden ist, Vinculin, Talin und paxillin [6]. Nach der Aktivierung rekrutiert FAK die Nicht-Rezeptor-Tyrosinkinase c-Src zu den focal adhesion Websites, um andere focal adhesion Proteine phosphoryliert wie paxillin und p130 Cas was zu Fokaladhäsionen (Abbildung 1B) reifen. Die Integrität und die Reifung von focal adhesion Komplexe Zyklus zwischen Montage an den Vorsprüngen und Demontage an der Hinterkante Zellen der Migration; jedoch sind die genauen molekularen Mechanismen sind nicht vollständig verstanden. In diesem Bericht fassen wir die aktuellen Erkenntnisse darüber, wie die Menschen krebserregend Helicobacter pylori
(H. pylori
) die Wirtszelle Aktinzytoskelett Stressfasern zu bilden und Adhäsionskomplexe dereguliert Veränderungen in der Zellform, die Migration zu induzieren und invasive Wachstum.
H. pylori
induziert Migration und invasive Wachstum von Magenepithelzellen
H. pylori
ist einer der erfolgreichsten menschlichen Erreger der Magenschleimhaut Epithel im Magen von etwa 50% der Bevölkerung der Welt kolonisiert. Sobald ausgerottet erworben und nicht durch Antibiotika, H. pylori
normalerweise im gesamten Lebensdauer bestehen bleibt, da der Host nicht in der Lage ist, die Infektion zu löschen. Nur eine Minderheit von 10-15% der infizierten Personen entwickelt Magenerkrankungen schwerer, die in erster Linie auf die bakterielle ausgedrückt pathogenen und Virulenz Faktoren abhängen, Umweltfaktoren und individueller genetischer Prädispositionen (zB
. Polymorphismen von Wirtsgenen wie Interleukin-1β (IL 1β), IL-8, IL-10, runt-verwandtes Gen 3 (RUNX3), etc
.), die Magen-Atrophie und Karzinogenese beeinflussen [10-12]. Die meisten schweren Komplikationen sind entzündliche Erkrankungen mit akuten und chronischen Gastritis oder Geschwüre des Magens und des Zwölffingerdarms, die schließlich in Malt (MALT) Lymphom und Magenkrebs [13] führen kann. Nach seiner Fähigkeit, Krebs zu fördern, H. pylori und Videos der Weltgesundheitsorganisation als Klasse-I Karzinogen [14]
H. eingestuft wurde. pylori
Pathogenese ist abhängig von der Expression von bakteriellen Virulenzfaktoren [10], die komplexe zelluläre Antworten von Magenepithelzellen beinhalten könnten [15, 16]. Die Vakuolen cytotoxin A (VacA) wird von vielen sezerniert wird, wenn nicht alle, H. pylori
Isolate und könnte die H. pylori
Virulenz obwohl seine pleiotropen Funktionen in vivo
verbessern. VacA bindet an viele Oberflächen Faktoren, einschließlich der rezeptorartigen Proteintyrosinphosphatase alpha und beta (RPTPα und RPTPβ) auf Wirtszellen präsentiert und nach Aufnahme induziert Membran Anion selektive Kanäle und Porenbildung, Apoptose und gigantischen Vakuolen in Wirtszellen [ ,,,0],17]. VacA ist ferner mit der Hemmung von T-Zell-Funktion assoziiert mit dem β2-Integrin-Rezeptors durch Bindung [18, 19]. Ein weiterer wichtiger pathogener Faktor ist das Cytotoxin-assoziierte Gen A (CagA), der viel Aufmerksamkeit auf sich gezogen hat, da seine Expression eng mit der Entwicklung schwerer Erkrankungen in vivo
[20, 21] verbunden ist. Die cagA
Gens innerhalb des cag
Pathogenitätsinsel (cag
PAI) Bereich auf dem Bakterienchromosom befindet, die Proteine wichtig für Aufbau und die Funktion eines speziellen Typ IV-Sekretionssystem (T4SS) [22 codiert, 23]. Wichtig ist gezeigt, wurde gefunden, daß die PAI cag
Protein CAGL eine T4SS-Pilus-Adhesin assoziiert für α5β1-Integrin stellt auf der epithelialen Wirtszelloberfläche exprimiert. Bindung des Fibronektin-nachahmt Arg-Gly-Asp (RGD) -Motiv in der CAGL Molekül Integrin &bgr; 1 ist notwendig, CagA in das Wirts Zytoplasma [24, 25] zu translozieren. Viele Studien beschrieben, dass CagA-positive H. pylori
Stämme sind eng mit der Entwicklung von akuter Gastritis verbunden, präneoplastischen und neoplastischen Läsion [26-29]. Ursächlicher Zusammenhang zwischen CagA und die Bildung von Neoplasien wurden in der mongolischen Wüstenrennmäusen nachgewiesen [30, 31] und in einem transgenen Mausmodell, in dem CagA neoplastischen Transformationen in vivo
induziert [32].
Bei gesunden Menschen die Magenepithels stellt wirksame erste Barrieren gegen Pathogene, die durch koordinierte Regulation der epithelialen Zellform, Polarität dicht verschlossen ist, Zell-Zell- und Zell-Matrix-Adhäsionen. Damit einhergehend mit der Kolonisierung des Magenschleim, H. pylori
abbaut die epitheliale Barrierefunktion zu Entzündungsreaktionen und neoplastischen Veränderungen induzieren abhängig von H. pylori
Virulenzfaktoren [33]. Dies könnte durch eine Umlagerung des Aktin-Zytoskelett als zentraler Mechanismus in diesen Prozessen erleichtert werden. Unterstützt wird dieser Vorschlag, H. pylori
induziert die Bildung von Vorsprüngen und massive Stressfasern in kultivierten Magenepithelzellen durch den Verlust von epithelialen Morphologie begleitet Zellen und Zell-zu-Zelle zu einem Streu Phänotyp invasive mitogene führende Adhäsionen in vitro
[33, 34] erinnert an Wachstumsfaktor-induzierte Epithelial-mesenchymale Transition (EMT). Der EMT Phänotyp eine komplexe morphogenetic Programm erfordert durch Veränderung der Genexpression initiiert, den Verlust von typischen epithelialen Eigenschaften und die Zunahme von mesenchymalen Eigenschaften [35], die in H. pylori nachgewiesen werden konnte
-colonized Zellen [36]. Während EMT, verlieren Zellen ihre polar, epitheliale Natur und erwerben eine sehr beweglich, mesenchymale Morphologie. Grundsätzlich ist EMT durch die (i) Zerlegen von interzellulären Verbindungen definiert, (ii) die Reorganisation der Aktin-Zytoskelett von Zell-Zell- und Zell-Matrix-junctions in protrusive und invasive Pseudopodien Strukturen wie Aktin Stressfasern und Aktin-abhängige Vorsprung von Zell Pseudopodien, (iii) und eine Zunahme der Zellbeweglichkeit. In der Regel treten diese Prozesse im synchronen Mode, aber unabhängig voneinander [35]. Dementsprechend effiziente H. pylori
-vermittelte Zellmigration ist eine äußerst komplexe koordinierte Prozess, der durch die Verlängerung der Lamellipodien an der Vorderkante der Zelle, die Montage der neuen focal adhesion-Komplexe, die Sekretion von Proteasen initiiert wird Kontakte zu der zu degradieren ECM die Bildung von invadopodia, Entwicklung von kontraktilen Kräften und schließlich Demontage von fokalen Adhäsionen führt zu tail Ablösung unterstützen (Abbildung 2) [34, 37]. Abbildung 2 Modell Epithelzellen der Migration. Für eine effiziente Migration entwickeln Epithelzellen neuen Aktin-abhängige Vorsprünge, die über neu zusammen fokalen Adhäsionen (rot) an der Vorderkante mit dem ECM verbunden sind. Sekretion von Proteasen ECM abzubauen erforderlich, um den Vorsprung in den ECM zu erstrecken invadopodia zu bilden. Am Heck reifte Fokaladhäsionen (grau) zu zerlegen, die Bewegung des Zellkörpers in einer definierten Richtung zu erleichtern. Aktin-abhängigen Vorsprung von Pseudopodienoberflächenerweiterungen ist ein Schlüsselelement bei der EMT-Migration von H. pylori
-colonized Zellen. Pathogen H. pylori-Stämme
ein morphogenetic Programm in unterschiedlichen Magen-Epithelzellen Linien induzieren, die eng mit den Merkmalen des EMT ähnelt [36]. CagA-transfizierten Zellen dringen durch die extrazelluläre Matrix über
die Bildung von invasiven Scheinfüsschen [38] darauf hinweist, dass CagA EMT in Magenkrebs-Zellen induzieren könnten. Funktionell diese Strukturen nachahmen invasive Podosomen oder invadopodia, und zeigen eine ähnliche Abhängigkeit von Matrix-Metalloproteasen (MMPs) für die Invasion. Zur Unterstützung dieses Konzept nicht-invasive Podosomen wurden durch invasive invadopodia in EMT (Abbildung 2) [39].
Haftung basiert räumlich-zeitliche Orchestrierung von Aktin-Polymerisation gesteuerte invasive Strukturen nach und nach ersetzt gezeigt werden [40] ist ein Merkmal vieler physiologischer und pathologischer Ereignisse. Die Spieler in diesem Szenario sind mechano empfindliche Moleküle, die auch on-Integrin vermittelter abhängig von außen in Signalkaskaden und viele der gleichen Spieler (wie Ezrin, Abl, Src, etc
.) Beinhalten, die für H. erforderlichen pylori
-induzierte Zellinvasion in das benachbarte Gewebe [38, 41, 42]. Ein wesentliches Element, das invadopodia von fokalen Adhäsionen ist die Modulation der Zelle sekretorischen Maschinen und die Brenn Sekretion von ECM-abbauenden Matrix-Metalloproteasen (MMPs), die letztlich ermöglichen, die Verletzung von Gewebegrenzen [43] trennt. Die ultrastrukturelle Merkmale und intrazellulären Dynamik von H. pylori
-induzierte Pseudopodien sind immer noch schlecht definiert, sondern eine Zukunft Identifizierung dieser Strukturen als invadopodia bezogenen zellulären Vorsprünge wäre keine Überraschung sein.
Als Infektion mit CagA
-positiven Stämme von H. pylori
ist eng mit der Induktion von Adenokarzinom des Magens assoziiert die durch injizierte CagA entführten Ziele wahrscheinlich pseudopod Bildung und das Eindringen von infizierten motile Zellen steuert. Tatsächlich in vitro
Studien haben gezeigt, dass das Protein CagA 2 (Grb2) [44], die Kinase kann verknüpfen Abl und Src gebunden Adaptormolekül Wachstumsfaktor-Rezeptor bindet Signalkaskaden Expression MMP und invadopodium Bildung [45] und somit beitragen der zur ortsspezifischen Bildung Komplexe, die für die Zellmigration und invasives Wachstum signalisieren. Interessanterweise H. pylori
induziert die Expression von MMP-7 im Lamellipodien motiler Zellen, die auch von aktivierten RhoA und Rac [46] ausgelöst wurde, eine enge Verbindung zwischen den ECM-Abbau, invasive Wachstum und effiziente Zellmotilität hindeutet. Die kortikale Zytoskelett dient als Bindeglied zwischen der extrazellulären Umgebung und dem Zytoplasma und ist so positioniert, zellulären Signalrelais zu koordinieren. Es kommt also nicht verwunderlich, dass Zytoskelett-assoziierten kortikalen Proteine eine Schlüsselrolle in der H. pylori
-induzierte Zellmodulation haben. Die mucinähnliche Trans Glykoprotein podoplanin kann auch induzieren EMT, Zellmigration und invasive Wachstum durch die ERM Rekrutierung (Ezrin, Radixin, Moesin) -Familie Protein Ezrin, ein Organisator der kortikalen Zytoskeletts, an der Plasmamembran. Diese Wechselwirkung ist für die Aktivierung des RhoA /ROCK Weges durch podoplanin [47, 48]. In H. pylori
-infizierten Magenepithelzellen wird Ezrin dephosphoryliert, die in der Entwicklung und Metastasierung von H. pylori beteiligt sein könnten
induzierten Magenkrebs [49]. Ezrin die Doppelrolle als Aktin-Bindung und GTPase Gerüstprotein weiter identifiziert dieses molekularen Komplexes als ein zentrales Ziel für die Zytoskelett-Umlagerungen zu verstehen, die für die Migration und invasive Wachstum von infizierten Epithelzellen führen [49, 50].
In der Tat, die EMT -ähnlichen Phänotyp von H. pylori
infiziertem epithelialen Wirtszellen impliziert die Bildung von Vorsprüngen und Dehnung. Eher unglücklich, Begriffe wie "Streuung Phänotyp" oder "Kolibri-Phänotyp" im Zusammenhang mit H. pylori-Infektion
wurde weithin als Synonym mit "Zellverlängerung" oder "Zellmigration". Interessanterweise sind thesaurierend Daten darauf hinweist, dass zelluläre Dehnung und Beweglichkeit sind unterschiedlich von H. pylori geregelt
über unabhängige Signaltransduktionswege [51]. Konsequent beobachtet, ist die drastische Verlängerung von Wirtszellen streng abhängig von CagA-Injektion [52-54], während H. pylori
-induzierte Zellbeweglichkeit
PAI-abhängige CAG ist, aber weitgehend CagA unabhängig [42, 51 , 55]. Machen diese Beobachtungen komplexer Daten häufen sich, dass CagA und VacA Funktionen einander in einigen Assays antagonisieren. Gemäß einer Studie, die zeigt, dass spezifische VacA Varianten gehemmt CagA-abhängige Zellverlängerung, reduzierte CagA-VacA vermittelte Apoptose und umgekehrt
, unterstreicht die störenden Funktionen von pathogenen Faktoren, ausgedrückt durch H. pylori
[56, 57] . Weiterhin H. pylori
es ausdrücklich pathogene Faktoren differentiell mit Wirtszellen, was zur Unterbrechung des Magenepithel und Bestimmen des Ergebnisses von Magenerkrankungen interagieren könnten. Rapid Wirtszellverlängerung und Migration sind insbesondere evident in menschlichen Magenkrebszellen (zB AGS-Zellen) [53, 58, 59], Brustkrebszellen (zB MCF-7-Zellen) [42, 60] und einen Subtyp des canine kidney Zelllinie MDCK [55, 61]. Milder und weniger ausgeprägte Entwicklung dieses typischen Phänotyp wurde in Magen MKN-1, MKN-28 und HS-746T Zellen in frühen Phasen von H. pylori
Infektionen beobachtet [15]. In Bezug auf die Zelle morphologischen und junctional Veränderungen, nur wenige Berichte über primäre Magenepithelzellen sind verfügbar [62, 63]. Wichtig ist, Krüger et al. demonstriert -induzierte Motilität und das Wachstum von ex vivo
isolierten Magenzellen [63]
H. pylori. Der Zugang zu Primärzellen ist begrenzt; daher ist es wichtig
auch beobachteten zellulären molekularen Mechanismen in vivo zu untersuchen. Bisher ist es noch spekulativ, wenn Veränderungen der Zellmorphologie beitragen tatsächlich zu H. pylori
assoziierte Magenerkrankungen, auch diese Prozesse wahrscheinlich Einfluss Wirtsreaktionen während Jahrzehnten anhaltender H. pylori
Infektionen. Helicobacter pylori
induzierte Signaltransduktionswege zu einem deregulierten Aktinzytoskelett unabhängig von CagA führenden
Obwohl klar ist, dass H. pylori
Zytoskelett-Veränderungen in den Epithelzellen induziert auffällig, Kenntnis der Signaltransduktionswege ist selten. In Serum-ausgehungert Zellen, beide CagA-positive und CagA-negativen H. pylori-Stämme
die Bildung von Aktin-Filamenten und lamellipodialen Strukturen [64] impliziert die Aktivierung von Rho-GTPasen vermittelt. In der Tat, die Aktivierung von Rac1 und Cdc42 wurde H. pylori
infiziertem AGS-Zellen [65] demonstriert. Mikroinjektion von inaktiven Rac verhindert Aktinzytoskelett Umlagerungen in lamellipodialen Strukturen in H. pylori
-colonized Zellen [64]. Durch Transfektion von dominant-negativen und katalytisch aktiven cDNA-Konstrukte oder unter Verwendung von gut charakterisierten GTPase-Targeting-Toxine, Crk Adapterproteine, Rac1 und H-Ras, aber nicht RhoA oder Cdc42 als entscheidende Komponenten, die zu H. pylori
identifiziert -induzierte Zellverlängerung [66]. Übereinstimmend mit Aktin-Polymerisation in H. pylori
-infizierten Zellen [64], die Aktivierung von Rho-GTPasen erfolgt unabhängig von CagA Injektion, aber offensichtlich die T4SS Vorrichtung erforderlich [65]. Da CAGL als Adhäsin für α5β1-Integrine identifiziert wurde, das CagA-Injektion und β1-Integrin-Aktivierung an der Spitze des T4SS verziert erlaubt [24], ist es verlockend, zu spekulieren, dass CAGL auch ein vielversprechender Kandidat für die Stimulierung Rho-GTPase-Aktivierung stellt (Abbildung 3A). Diese Hypothese wird derzeit von der Feststellung gestützt, dass CAGL beschichteten Latexkügelchen Membran über
Integrin-vermittelte Aktivierung von FAK und Src [24] Kräuseln stimuliert. Ein weiteres mögliches Szenario schlägt ist OIPA als induzierende Faktor, da OIPA
Mutanten wurden weniger gemeldet FAK aktivieren vermutlich unabhängig von cag
PAI oder CagA [67]; jedoch Experimente mit recomplemented OIPA
Mutanten sind anhängig. Abbildung 3 Schematische Übersicht über CAGL und CagA-vermittelte Signaltransduktionswege in H. pylori beteiligt -induzierte Zellmotilität und Dehnung. (A) H. pylori
CAGL an der Spitze des T4SS drückt, der direkt Integrinen auf Magenepithelzellen präsentiert &bgr; 1 bindet. Aktivierte β1 Integrin stimuliert FAK und Src-Aktivität in frühen Phasen von H. pylori-Infektionen
. FAK phosphoryliert paxillin nach einer Infektion, die c-Abl-phosphoryliert Crk Signalisierung beitragen könnte, die von SFK-Aktivität über
paxillin oder p130CAS beeinflusst werden könnte. FAK, SFKs und Abl-Kinase-vermittelte Aktivierung von Crk Proteine können das Aktin-Zytoskelett durch die Zellmigration zu epithelialen beitragen DOCK180 /Rac1 /WAVE /Arp2 /3-Weg regulieren. (B) CAGL-Integrin-Bindung führt zur Translokation des pathogenen Faktor CagA in das Wirts Zytoplasma
H. pylori. CagA wird rasch von Kinasen der Src-Familie (SFK) und binden an eine Vielzahl von Wirtszellen Faktoren (X) in seiner phosphorylierten und nicht-phosphorylierten Form phosphoryliert. Tyrosinphosphoryliert CagA interagiert mit SHP-2 und Csk FAK und Src in späten Phasen der H.-pylori-Infektion
zu inaktivieren. Während inaktivierten Src durch aktivierte Abl-Kinasen ersetzt CagA-Phosphorylierung zu erhalten, führt inaktive Src Dephosphorylierung von Src Zielmoleküle Ezrin zu Tyrosin, Vinculin und Cortactin. Cortactin ist dann Serin phosphoryliert von H. pylori
-aktivkohle ERK1 /2-Kinasen, die Zellverlängerung entscheidend beiträgt. Schwarze Pfeile, -induzierten direkte Signalwege
H. pylori. Gepunktete Pfeile,
H. pylori-induzierten oder indirekten Signalwege-Src vermittelt. Graue Pfeile, Inaktivierung Signalwege. Roter Pfeil, CagA Injektion als zentrale Schritt in der Regulation der fokalen Adhäsionen. P, phosphorylierte Proteine. X, Wirtszellproteine.
Die β1 Integrin /FAK /Src Weg sendet Signale an das Aktin-Zytoskelett über
paxillin, ein wichtiges Gerüstprotein in fokalen Adhäsionen liegt [34]. In H. pylori
infiziertem Zellen aktiviert FAK phosphoryliert Tyrosin 118 im paxillin Protein (paxillin Y118), die für die Zellbewegung als Reaktion auf H. pylori
[68] wesentlich war. Da phosphorylierten paxillin Y118 bindet das Adaptor-Protein v-CRK-Sarkom-Virus CT10 Onkogen Homolog (Crk) als Reaktion auf Zellhaftung, platelet-derived growth factor (PDGF) oder Angiotensin II [69], H. pylori
- ausgelöst paxillin Y118 Phosphorylierung auch vor der Aktivierung von Crk /DOCK180 (dedicator von Zytokinese) wirken kann /Rac1 /WAVE /Arp2 /3-Signaltransduktionsweg in H. pylori
infiziertem Zellen, die erfasst wurde, Eine andere Studie (3A) [70]. Alternativ H. pylori
-induzierte Src-Aktivität konnte aktivieren p130 Cas auf die Rekrutierung des CRK-Komplexes führt; jedoch eine Beteiligung von p130 Cas in H. pylori
-vermittelte Zytoskelett-Umlagerung noch nachgewiesen werden muss (Abbildung 3A).
Verordnung von CagA-vermittelten Wirtszelle Dehnung
die H. pylori
-induzierten Veränderungen der Zellmorphologie werden durch die drastische Dehnung von Epithelzellen dominiert, die sowohl das Aktin-Zytoskelett und fokale Adhäsionen aktive Regelung beinhaltet. Einzellige Analysen vorgeschlagen, dass H. pylori
-abhängigen Zellverlängerung könnte durch deregulierte Fokaladhäsionen statt Aktinzytoskelett Umlagerung vermittelt werden. Stabilisierte fokalen Adhäsionen verursachen einen Defekt in der Zell Retraktion was zur Bildung von starken Zugkräften auf motile H. pylori
-infizierten Zellen [52]. CagA erhöht Phosphorylierung und anschließende Aktivierung von Myosin Light Chain (MLC) in einem Drosophila-Modell [71]. Die gleichzeitige mispatterning von MLC führt zu Zellverlängerung aufgrund Rückzug Versagen und die Störung der epithelialen Morphologie und Integrität. Basierend auf einer Phospho-Proteomanalyse der Aktin-bindenden Protein gefäßerweiternden-stimulierte Phosphoprotein (VASP) identifiziert wurde, die co-lokalisiert mit fokalen Adhäsionen von H. pylori
infiziertem Zellen [72]. Die Herunterregulierung von VASP-Expression und die Hemmung der VASP-Phosphorylierung blockiert Zellverlängerung in Reaktion auf H. pylori
, aber es wurde nicht untersucht, ob phosphoryliert VASP die Zerlegung von fokalen Adhäsionen gestört [72].
Die Bedeutung von fokalen Adhäsionen bei der Förderung in den Prozess der Zellverlängerung [24] Zellverlängerung wurde, ist wichtig, durch die Feststellung, dass β1-Integrin-vermittelten Injektion von CagA betont. Bei der Translokation, lokalisiert CagA an der inneren Membran der infizierten Zellen, wo sie sich rasch von den Nicht-Rezeptor-Tyrosinkinasen c-Src, Fyn, Lyn und Ja der Src-Familie-Kinasen (SFK) phosphoryliert wird [73, 74]. Phosphorylierungsstellen wurden in einem Glu-Pro-Ile-Tyr-Ala-Sequenz (Epiya Motiv) lokalisiert, die 1-5 Wiederholungen wie andere existiert, nämlich Epiya-A, Epiya-B, Epiya-C in West H. pylori
isoliert und Epiya-B, Epiya-D in ostasiatische Stämme [75, 76] Epiya-A. Die Src-vermittelte CagA Phosphorylierung (CagA PY) durch eine schnelle Inaktivierung von Src-Kinase-Aktivität, ausgelöst durch die Bindung von CagA an die C-terminale Src-Kinase (Csk) (3B) [54, 58] gefolgt. Src-Kinase-Inaktivierung führt dann zur Dephosphorylierung von Src Zielproteine wie Vinculin, Ezrin und Cortactin [49, 54, 77]. In der Tat, die Tyrosin-Phosphorylierung von CagA PY zusammen mit der Dephosphorylierung von SFKs und ihre Zielmoleküle sind wichtig in den Prozess der Regulierung der Aktin-Zytoskelett und fokalen Adhäsionen, die mit den drastischen morphologischen Veränderungen von H. pylori trägt
- infizierten Zellen (3B).
Ein weiteres Schlüsselmolekül in der H. pylori
stimulierte Zellverlängerung ist SHP-2 (src-Homologie 2 Domain-Tyrosin-Phosphatase) (3B). Analyse von ektopisch exprimiert CagA und isogenen Phosphorylierung-resistente Mutanten zeigten, dass CagA PY bindet direkt an SHP-2, die von SHP-2 zu einer Erhöhung der Phosphatase-Aktivität führte [78, 79]. Die CagA /SHP-2-Komplex wurde ebenfalls in der Magenschleimhaut von H. pylori
-positiven Patienten mit Gastritis und Frühstadien von Magenkrebs [80] festgestellt. Die Aktivierung von SHP-2-Phosphatase-Aktivität wurde folglich berichtet FAK in Zellen zu inaktivieren, die ektopisch CagA exprimieren [81]. Im Gegensatz zu aktiviertem FAK kann dephosphoryliert FAK nicht in fokalen Adhäsionen lokalisiert werden, die die Entwicklung des länglichen Zellphänotyp unterstützen könnten. Im Gegensatz zu dieser Beobachtung CAGL und OIPA aktivieren FAK in H. pylori
infiziertem Zellen [24, 67]. Vor kurzem wurde eine neue funktionelle Form Cortactin berichtet, weiter unterstreicht die Bedeutung der Cortactin als kritischer Mediator in Signaltransduktionswegen in H. pylori
infizierten Wirtszellen (3B). Nach Tyrosindephosphorylierung-Src vermittelt wird, wird Cortactin phosphoryliert an Serin 405 (Cortactin S405). Phosphorylated Cortactin S405 bindet stark an und aktiviert FAK. Cortactin S405 Phosphorylierung wurde von ERK1 /2-Kinasen vermittelt und Macht Falle aktiviert FAK zu einem gestörten Umsatz von fokalen Adhäsionen führen (3B) [82]. Dies ist einer der ersten identifizierten Mechanismen erklären, warum die Aktivierung von mitogen-aktivierten Proteinkinasen (MAP) über Rap1 GTPasen [83] oder Proteinkinasen C (PKC) [84] in Antwort auf H. pylori-Infektionen zu
Zelle beitragen Dehnung [61, 70, 82]. dephosphoryliert SFK Zielmoleküle, die Phosphorylierung von CagA PY ist potent nachhaltig durch aktivierte Abl-Kinasen nach der Inaktivierung von Src [60, 85] Im Gegensatz zu
. Abl-Kinasen halten CagA PY-Phosphorylierung und CagA PY-abhängige Downstream-Effekte, die noch nicht vollständig verstanden. Interessanterweise wurde darauf hingewiesen, dass CagA East Asian-Typ induziert deutlich stärkere Wirkungen auf Ratten-Zellwachstum als die westliche CagA [86] transfiziert, die zu den verschiedenen Epiya Motive offensichtlich zurückzuführen sind und ihre Bindungsaffinitäten zu SHP-2 [75]. Da es nicht klar ist, sind, wenn Src und Abl-Kinasen bevorzugen unterschiedliche Epiya Motive oder weisen ähnliche Phosphorylierung Affinitäten, weitere Analysen notwendig, die SFK und Abl-Kinase-vermittelten CagA-Phosphorylierung zu untersuchen. Activated c-Abl
folglich phosphoryliert auch Adapterproteine Crk [ ,,,0],60, 85], die mit CagA PY [70] verbindet eine große CagA PY rekrutiert Proteinkomplex mit Signaltransduktionswege in Richtung des Aktin-Zytoskelett (3B) berichtet wurde, zu interagieren. Diverse, aber koordinierte Signaltransduktionswege konvergieren auf CagA PY als wichtige zentrale Schlüsselmolekül in der H. pylori
vermittelter Zellmigration [76]. Neben SHP-2 als die erste identifizierte Bindungspartner von CagA [78], viele andere Bindungspartner für phosphorylierten und nicht-phosphorylierten CagA haben in den letzten Jahren einschließlich Par1 /MARK, c-Met, PLCy (Phospholipase C gamma) identifiziert worden ist, ZO-1 (Zonula Occludens-1), Csk (c-Src-Tyrosinkinase
) Gab1 (GRB-assoziiertes Bindemittel 1
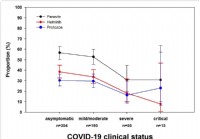 Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
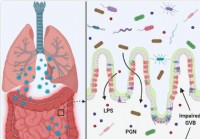 Leaky Gut und mikrobielle Dysbiose könnten bei schwerkranken COVID-19-Fällen zum Zytokinsturm beitragen
Leaky Gut und mikrobielle Dysbiose könnten bei schwerkranken COVID-19-Fällen zum Zytokinsturm beitragen
 Die langfristige Anwendung von Antibiotika bei Frühgeborenen fördert arzneimittelresistente Darmbakterien
Die langfristige Anwendung von Antibiotika bei Frühgeborenen fördert arzneimittelresistente Darmbakterien
 Anti-Coronavirus-Moleküle von Mikroben könnten der Schlüssel zu neuen Behandlungen sein
Anti-Coronavirus-Moleküle von Mikroben könnten der Schlüssel zu neuen Behandlungen sein
 Männer, die zweimal pro Woche Joghurt essen, erkranken seltener an Darmkrebs
Männer, die zweimal pro Woche Joghurt essen, erkranken seltener an Darmkrebs
 Pflanzliche Lebensmittel können antibiotikaresistente Superbakterien auf den Menschen übertragen
Pflanzliche Lebensmittel können antibiotikaresistente Superbakterien auf den Menschen übertragen
 Studie deutet auf Zusammenhang zwischen probiotischem Konsum und „Gehirnnebel“ hin
Eine am Medical College of Georgia an der Augusta University durchgeführte Studie hat gezeigt, dass die Einnahme von Probiotika zu einer signifikanten Ansammlung von Dünndarmbakterien führen kann, die
Studie deutet auf Zusammenhang zwischen probiotischem Konsum und „Gehirnnebel“ hin
Eine am Medical College of Georgia an der Augusta University durchgeführte Studie hat gezeigt, dass die Einnahme von Probiotika zu einer signifikanten Ansammlung von Dünndarmbakterien führen kann, die
 Die westliche Ernährung kann das Risiko einer „tödlichen Sepsis“ erhöhen.
Experten warnen Neue Forschungen, die an der Portland State University durchgeführt wurden, legen nahe, dass die westliche Ernährung das Risiko einer schweren Sepsis und der Sterblichkeit durch die I
Die westliche Ernährung kann das Risiko einer „tödlichen Sepsis“ erhöhen.
Experten warnen Neue Forschungen, die an der Portland State University durchgeführt wurden, legen nahe, dass die westliche Ernährung das Risiko einer schweren Sepsis und der Sterblichkeit durch die I
 Forschung sagt bei SARS-CoV-2-Infektion des Hundes,
Übertragung unwahrscheinlich Eine neue Studie auf dem Preprint-Server veröffentlicht bioRxiv* im September 2020 zeigt, dass Hunde an einer mysteriösen Atemwegsinfektion erkrankt sind und sterben. ze
Forschung sagt bei SARS-CoV-2-Infektion des Hundes,
Übertragung unwahrscheinlich Eine neue Studie auf dem Preprint-Server veröffentlicht bioRxiv* im September 2020 zeigt, dass Hunde an einer mysteriösen Atemwegsinfektion erkrankt sind und sterben. ze