Genomic caractérisation d'un Helicobacter pylori
isoler un patient avec un cancer gastrique Résumé
Contexte de la Chine
Helicobacter pylori est
bien connu pour ses relations avec l'apparition de plusieurs maladies gastriques graves. Les mécanismes de la pathogenèse déclenchée par H. pylori
sont moins bien connus. Dans cette étude, nous rapportons la séquence du génome et des caractérisations génomiques de H. pylori de la souche HLJ039 qui a été isolé à partir d'un patient avec un cancer gastrique dans la province chinoise du Heilongjiang, où il y a une forte incidence du cancer gastrique. Pour étudier les caractéristiques génomiques potentielles qui peuvent être impliqués dans la pathogenèse du carcinome, le génome a été comparé à trois génomes déjà séquencés dans ce domaine.
Résultat
Nous avons obtenu 42 contigs avec une longueur totale de 1.611.192 pb et prédit 1,687 séquences codantes . Par rapport à des souches isolées à partir de la gastrite et les ulcères dans ce domaine, 10 régions différentes ont été identifiées comme étant unique pour HLJ039; ils encodés principalement de type II de modification de restriction enzyme, type II M6a méthylase, ADN méthyltransférase-cytosine, l'ADN méthylase, et des protéines hypothétiques. Un partage de l'identité de 93% avec une protéine hypothétique de Helicobacter cinaedi
ATCC BAA-847 unique, fragment 547 pb n'a pas été présent dans toutes les autres souches de H. pylori
précédentes. analyse phylogénétique basée sur le génome central polymorphismes de nucléotides simples montre que HLJ039 est défini comme hspEAsia sous-groupe, qui appartient au groupe hpEastAsia.
Conclusion
méthylations de l'ADN, les variations des régions génomiques impliquées dans les systèmes de restriction et de modification, sont " régions chaudes "qui peuvent être liés au mécanisme du cancer gastrique induite par H. pylori. La séquence du génome fournira des informations utiles pour l'exploitation minière en profondeur des mécanismes potentiels liés au cancer gastrique Asie de l'Est.
Mots-clés
Helicobacter pylori
cancer gastrique séquençage de nouvelle génération Genomic dispose Contexte
Helicobacter pylori
, une bactérie à Gram négatif qui colonise l'estomac humain, a été largement reconnu comme étant une bactérie pathogène liées à la pathogenèse de la gastrite, les ulcères et le carcinome [1-3]. La variabilité génétique de H. pylori
entraîne sa capacité dramatique à adapter à la niche gastrique [4-9]. Cependant, bien que de nombreuses études ont été réalisées, ses mécanismes ne sont pas encore bien élucidés.
Avec le développement rapide de la technologie de séquençage de prochaine génération et la réduction des coûts, il est devenu possible d'effectuer de grandes procédures de séquençage du génome à l'échelle pour obtenir de nombreuses informations sur structure de la population et les maladies des marqueurs biologiques. Au cours des dernières années, de plus en plus de souches
H. pylori provenant de différentes régions géographiques, les ethnies et les maladies ont été séquencés [10-12], et au moins 50 séquences génomiques sont actuellement disponibles dans les bases de données publiques.
Dans une étude précédente, nous avons publié des séquences du génome de trois souches récupérées chez des patients souffrant d'ulcères et la gastrite atrophique dans la province du Heilongjiang [13]. Il est bien connu que les souches de H. pylori isolées à partir de différentes zones géographiques montrent la diversité génomique dramatique [14]. Ainsi, au niveau génomique, l'analyse comparative entre les souches de différentes manifestations cliniques doit d'abord éliminer ces brouillages. Comparative génomique analyse de séquençage des souches isolées chez les patients célibataires pourrait être un moyen fiable pour éliminer ces brouillages [15-17]. Cependant, il est généralement difficile de suivre un patient et obtenir des souches isolées à partir de diverses manifestations imprévisibles.
Dans cette étude, nous avons rapporté un projet de séquençage du génome de la souche HLJ039 qui a été isolé à partir d'un patient avec un cancer gastrique dans la province de Heilongjiang. Après l'intégration avec les trois autres génomes de la même région, l'analyse génomique comparative initiale a été réalisée pour étudier les caractéristiques génétiques du cancer gastrique isolats.
Méthodes
Strain sélection
HLJ039 a été isolé à partir d'un 84-year-old homme différencié cancer du corps de l'estomac mal. Bien que d'autres souches de H. pylori
liées carcinome gastrique isolées de différentes régions, ethnies et populations dans le monde sont présentes dans les bases de données publiques, nous ne sélectionnons ces souches pour notre analyse comparative. La souche de fond complexe, il sera très difficile d'identifier les caractéristiques génomiques fiables qui peuvent être contribué à une maladie spécifique comme le cancer gastrique. En tant que tel, l'analyse d'une région géographique, l'appartenance ethnique, ou d'une population spécifique peut être un moyen plus judicieux de trouver des indices potentiels liés à des maladies spécifiques. Par conséquent, dans cette étude, nous avons sélectionné trois souches isolées de la province du Heilongjiang pour l'analyse comparative. Ces souches sont très représentatives parce que la province du Heilongjiang a une incidence élevée de maladies gastriques en Chine, en particulier pour le cancer gastrique. En outre, la province du Heilongjiang chinois est proche de la Corée et le Japon. Ces pays d'Asie orientale ont censément la plus forte incidence du cancer gastrique dans le monde entier [18, 19] l'approbation éthique de.
Cette recherche a été approuvé par la réunion du comité d'éthique de l'Institut national de lutte contre les maladies transmissibles et la prévention, la Chine CDC, selon l'éthique des lois et règlements chinois. NO:. ICDC-2013001
séquençage du génome et l'annotation
La souche a été isolée de la muqueuse gastrique et cultivée sur Columbia base de gélose au sang de mouton à 5%. L'ADN a été extrait comme décrit précédemment [20]. Pour chaque souche, le séquençage du génome entier a été réalisée en utilisant un Illumina HiSeq 2000 par génération de bibliothèques jumelé de gamme (500 pb et 2 kb) suivant les instructions du fabricant. Les longueurs de lecture étaient de 90 pb et 50 pb pour chaque bibliothèque, à partir de laquelle plus de 100 Mo de données de haute qualité a été généré. La fin appariés lit des deux bibliothèques étaient de novo assemblés en échafauds utilisant SOAPdenovo (http:... //Savon génomique org cn). la prédiction de gène a été réalisée à l'aide Glimmer. Les gènes d'ARNt ont été recherchées par tRNAScan-SE2, tandis que les gènes ARNr ont été recherchées par RNAmmer3. Protein BLAST4 a été exécuté en utilisant les séquences codantes traduites comme une requête contre la séquence de référence (H. pylori
souche 51).
Le génome a encore été annotée et fonctionnellement catégorisée par Rapid Annotation à l'aide du sous-système de la technologie (RAST). Un sous-système est un ensemble de rôles fonctionnels qu'un annotateur a décidé sont liées. Subsystems représentent souvent la perception des rôles fonctionnels qui composent une voie métabolique, complexe, ou classe de protéines [21] Première analyse comparative génomique et phylogénétique
. Pour identifier les régions qui pourraient être impliqués dans la pathogenèse du cancer gastrique, MAUVE a été utilisé pour comparer les HLJ039 avec trois isolats supplémentaires récupérés de la même région [22]. Comme décrit précédemment, HLJ271 a été récupéré chez un patient présentant un ulcère gastrique. HLJ193 et HLJ256 ont été récupérés des patients souffrant de gastrite atrophique. Différentes régions (DRS) de HLJ039 ont été marqués le long de son emplacement chromosomique. DRs se réfèrent à une séquence codante (CDS), insertion et deletion dans HLJ039 par rapport aux trois autres génomes.
Pour définir la caractérisation phylogénétique des HLJ039 en utilisant la disposition du public de H. pylori
séquences génomiques, des séquences 53 génomiques entières ont été extraites GenBank pour la construction de l'arbre phylogénétique (fichier supplémentaire 1). P12 a été utilisé comme un génome de référence. Les comparaisons ont été effectuées à l'aide du programme nucmer de MUMMER3 mis en œuvre dans Panseq [23]. Génomes ont été fragmentés en segments de 500 pb qui devaient être présents dans les 54 génomes être inclus dans le génome du noyau. Horizontalement gènes transférés ont généralement une grande diversité génétique entre les différentes souches, par exemple, les zones de plasticité, quels systèmes de type encode IV de sécrétion, les systèmes R-M, ou îlots génomiques transférables. Selon le principe de l'alignement multiple par l'utilisation d'Panseq, ces gènes horizontaux potentiels seraient éliminés des gènes essentiels. polymorphismes nucléotidiques simples (SNP) dans les génomes de base sont déterminés et utilisés pour générer un fichier Phylip-formaté. SNPs concaténés longueur de 29259 pb ont été utilisés pour construire un arbre phylogénétique en utilisant le procédé de neighbor-joining dans MEGA5. méthode Bootstrap a été utilisé pour évaluer la stabilité des relations phylogénétiques.
dépôt de données génomiques
L'ensemble de ce projet génome de fusil de chasse a été déposé à DDBJ /EMBL /GenBank sous le numéro d'JAAA00000000, tandis que la version JAAA01000000 est décrite dans le présent document. assurance de la qualité
L'ADN génomique a été extrait à partir d'une souche cultivée H. pylori pur
et confirmé en utilisant des tests biochimiques classiques (positifs pour l'uréase, la catalase et oxydase). Le serveur RAST a été utilisé pour évaluer les contaminations hétérogènes potentiels.
Les premiers résultats
Nous avons finalement obtenus 42 contigs avec une longueur totale de 1.611.192 pb et prédit 1,687 CDS dans le projet de génome de la souche HLJ039. Des informations supplémentaires sont incluses dans les rapports de séquençage de HLJ039 (fichier supplémentaire 2). La teneur en G + C est 38,72%. La distribution du sous-système et des informations générales sur la répartition fonctionnelle potentielle de HLJ039 sont présentés dans la figure 1. Par rapport aux trois génomes HLJ supplémentaires, HLJ039 dispose de 10 régions différentes (DRS). Des informations détaillées sur ces fragments est présenté dans le tableau 1. Les emplacements de ces projets de résolution sont marqués dans l'ensemble du génome (Figure 2). Environ la moitié de ces séquences encodées protéines hypothétiques. La plupart des séquences DR protéines impliquées dans la méthylase d'ADN et une enzyme de modification de restriction codées. Notamment, un fragment de 547 pb uniques (DR9) partageant 93% d'identité avec une protéine hypothétique de Helicobacter cinaedi
ATCC BAA-847 a été trouvé qui n'a jamais été présent dans toutes les autres souches de H. pylori
précédemment, qui indique un transfert horizontal de gènes possible entre H. pylori et H.
cinaedi
. DR9, situé dans le squelette 5, insère dans un gène codant 1371 pb de type III endonucléase de restriction, qui est responsable de modifications de l'ADN méthylase spécifique de l'adénine. Figure 1 statistiques de distribution du sous-système de Helicobacter pylori souche HLJ039 générée par l'annotation rapide en utilisant le serveur de la technologie du sous-système.
Tableau 1 Généralités sur les différentes régions (drs) dans HLJ039 de DR au
Début de
End
Gene Description
DR1
145736
180926
25 protéines hypothétiques, VirB4, l'ADN topoisomérase I, Pará, protéine élément mobile, premier ORF en transposon ISC1904
DR2
618.752
619.703
fucosyltransférase
DR3
740.131
740.654
protéines hypothétique
DR4
1200420
1202309
protéines hypothétique
ADN méthyltransférase-cytosine
DR5
1254233
1256053
protéines hypothétique
DR6
1335551 1337398
type II M6a méthylase (hinFIM)
hypAIVR
DR7
1393932
1394805
protéines hypothétique
DR8
1443251 1445196
type enzyme de modification de l'ADN II
protéine hypothétique
DR9
1484058
1484604
hypothétique partageant 93% d'identité avec un fragment de Helicobacter cinaedi protéines
ATCC BAA-847
DR10
1538060
1539662
restriction Type IIG et modification enzymatique
Figure 2 alignement du génome du carcinome gastrique isoler HLJ039 avec des non-carcinome isolats.
Tous les résultats ci-dessus mettent en évidence le rôle important des systèmes de modification de restriction d'ADN chez H. pylori de recombinaison génomique. Un total de 29,259 SNP de base ont été trouvés parmi les 54 séquences génomiques analysées. Basé sur une analyse du génome central de SNP de 54H. Les souches pylori de réparties dans différentes régions du monde, un arbre phylogénétique a été généré pour montrer le sous-type HLJ039. Toutes les souches ont été classés en différents groupes définis par des études antérieures selon multilocus sequence typing [24, 25]. La figure 3 montre que HLJ039 a été défini comme appartenant au sous-groupe hspEAsia, qui appartenait au groupe hpEastAsia. Figure 3 L'analyse phylogénétique des 54 souches Helicobacter pylori en fonction de leur génome de base polymorphismes nucléotidiques.
Note: Les différentes régions (DRS) fait référence au codage insertion de la séquence et la suppression dans HLJ039 par rapport aux trois autres génomes
Orientations futures
L'incidence du cancer gastrique dans les pays d'Asie orientale est assez élevé [18, 19. ]. Pour explorer les mécanismes pathogènes potentiels qui peuvent contribuer à ce phénomène, plus asiatique H. pylori
souches doivent d'abord être séquencés. Les souches sélectionnées pour le séquençage doivent être représentatifs et éliminer la variation géographique. Nos orientations futures seront axées sur le séquençage génomique à grande échelle de différents isolats cliniques provenant de zones avec une incidence élevée de cancer gastrique. Des analyses plus détaillées impliquées dans la méthylation de l'ADN ainsi que des systèmes de restriction et de modification seraient les directions les plus attrayantes pour les études de H. pylori
cancer gastrique induite.
Consentement
Le consentement éclairé écrit a été obtenu à partir du patient pour la publication de ce rapport et les images qui les accompagnent.
disponibilité des données à l'appui
données supplémentaires à l'appui des résultats présentés ici sont inclus dans les fichiers supplémentaires
Déclarations Remerciements.
ce travail a été soutenu par un fonds pour la Chine Mega-projet pour les maladies infectieuses (2011ZX10004-001) et une subvention de la national Technology R &. Programme D dans le 12e plan quinquennal de la Chine (2012BAI06B02)
matériel supplémentaire électronique
13099_2014_126_MOESM1_ESM. doc fichiers supplémentaires 1: informations générales pour les génomes disponibles publiquement (dOC 60 Ko) 13099_2014_126_MOESM2_ESM.doc fichiers supplémentaires 2:.. informations Assemblée pour HLJ039 (dOC 27 Ko) Auteurs «original soumis fichiers pour les images
Voici les liens vers l'origine des auteurs ont soumis des dossiers pour les images. de fichier d'origine pour la figure 1 13099_2014_126_MOESM4_ESM.tiff Auteurs 13099_2014_126_MOESM3_ESM.tiff Auteurs fichier d'origine pour la figure 2 13099_2014_126_MOESM5_ESM.tiff Auteurs fichier d'origine pour la figure 3 Intérêts concurrents
Les auteurs déclarent qu'ils ont aucun conflit d'intérêts.
Auteurs ' contributions
YY effectué l'analyse bioinformatique et a écrit le manuscrit; MZ et LH étaient responsables de l'isolement de bactéries et d'identification; LL xh et YZ effectué le séquençage génomique; JZ et PN conçus l'étude et ont fourni un soutien financier à ce travail. Tous les auteurs ont lu et approuvé le manuscrit final.
 La metformine pourrait aider les intestins qui fuient
La metformine pourrait aider les intestins qui fuient
 La recherche montre comment les microbes intestinaux affectent la grippe intestinale
La recherche montre comment les microbes intestinaux affectent la grippe intestinale
 La découverte de 100 nouveaux gènes pourrait aider la recherche sur les maladies de la pigmentation
La découverte de 100 nouveaux gènes pourrait aider la recherche sur les maladies de la pigmentation
 Les pommes biologiques ont des propriétés probiotiques
Les pommes biologiques ont des propriétés probiotiques
 Les aliments végétaux peuvent transmettre des superbactéries résistantes aux antibiotiques aux humains
Les aliments végétaux peuvent transmettre des superbactéries résistantes aux antibiotiques aux humains
 L'acide glycyrrhizique comme candidat médicament pour COVID-19
L'acide glycyrrhizique comme candidat médicament pour COVID-19
 La maladie de Crohn
La maladie de Crohn provoque une inflammation du tractus gastro-intestinal. Elle peut être confondue avec la rectocolite hémorragique et le syndrome du côlon irritable, mais la maladie de Crohn est un
La maladie de Crohn
La maladie de Crohn provoque une inflammation du tractus gastro-intestinal. Elle peut être confondue avec la rectocolite hémorragique et le syndrome du côlon irritable, mais la maladie de Crohn est un
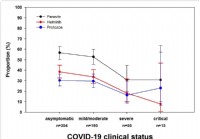 La recherche montre que les infestations de parasites intestinaux réduisent la gravité du COVID-19
Nous en apprenons chaque jour davantage sur la maladie COVID-19. Les adultes de tout âge atteints de certaines conditions médicales sous-jacentes courent un risque accru de maladie grave due au virus
La recherche montre que les infestations de parasites intestinaux réduisent la gravité du COVID-19
Nous en apprenons chaque jour davantage sur la maladie COVID-19. Les adultes de tout âge atteints de certaines conditions médicales sous-jacentes courent un risque accru de maladie grave due au virus
 La recherche dit que dans l'infection canine par le SRAS-CoV-2,
transmission peu probable Une nouvelle étude publiée sur le serveur de préimpression bioRxiv* en septembre 2020 montre que les chiens de compagnie tombent malades et meurent dune mystérieuse infecti
La recherche dit que dans l'infection canine par le SRAS-CoV-2,
transmission peu probable Une nouvelle étude publiée sur le serveur de préimpression bioRxiv* en septembre 2020 montre que les chiens de compagnie tombent malades et meurent dune mystérieuse infecti