análise do perfil genômico de cancros gástricos de tipo difuso da arte abstracta
Fundo
O câncer de estômago é o terceiro mais mortal entre todos os tipos de câncer em todo o mundo. Embora a incidência de cancro gástrico do tipo intestinal diminuiu, a incidência de difuso de tipo continua a aumentar e a sua progressão é notoriamente agressivo. Não existe informação suficiente sobre as variações do genoma do câncer gástrico tipo difuso, porque suas células são normalmente misturado com células normais, e esta baixa celularidade fez difícil analisar o genoma.
Resultados
Analisamos genomas inteiros e exomes correspondentes de câncer gástrico tipo difuso, usando tumor combinados e amostras normais de 14 difusa do tipo e cinco pacientes com câncer gástrico do tipo intestinal. Somatic variações encontradas no cancro gástrico do tipo difuso são comparados com aqueles do tipo intestinal e relatado anteriormente variantes. Nós determinamos a taxa somática exônico média mutação dos dois tipos. Encontramos genes associados do controlador de candidato, e identificar sete novas mutações somáticas no CDH1
, que é um gene associado a um cancro gástrico bem conhecido. a análise da estrutura tridimensional da proteína E-caderina mutada sugere que estas novas mutações somáticas podem causar perturbações funcionais significativas de locais críticos de ligação ao cálcio na junção EC1-2. A análise mostra que a instabilidade cromossómica do gene MDM2
é amplificado. Após análise estrutural completa, um TSC2 novel gene de fusão
-RNF216
é identificado, o que pode, simultaneamente, perturbar percursos tumor-supressora e ativar tumorigênese.
Conclusões
Relatamos o perfil genômico de tipo difuso gástrica cancros, incluindo novas variações somáticas, um gene de fusão romance, e amplificação e supressão de determinadas regiões cromossômicas que contêm oncogenes e supressores tumorais.
Fundo
fileiras de câncer de estômago como a terceira causa mais importante de mortalidade por câncer mundial [1]. Histopatologicamente, cancro gástrico (GC) podem ser classificados em duas categorias com base em diferenças morfológicas: tipo intestinal GC (CIG) e do tipo difuso GC (DGC) [2, 3]. IGC é tipicamente associada com a infecção por Helicobacter pylori
, e é especialmente comum no Japão e Coreia do [4-6]. DGC é uniformemente distribuída geograficamente, e inclui formas agressivas clínicas, como linite plástica, que têm mau prognóstico, especialmente em pacientes jovens [7, 8]. modificações de DNA genômico que conduzem a GC pode acontecer como resultado de vários fatores de risco ambiental, tais como uma dieta rica em sal e fumo de tabaco [9]. Embora a incidência de IGC diminuiu de forma constante ao longo de várias décadas (44% de redução, de 1978 a 2005), DGC aumentou rapidamente (em 62%) a partir de 1978 até 2000, antes de diminuir ligeiramente no período 2001-2005 [10]. Apesar da evidência cumulativa que IGC e DGC desenvolvem através de diferentes vias cancerígenos [11, 12], dados detalhados escala genômica para DGC estão faltando por causa da disponibilidade limitada de amostras clínicas e um baixo nível de pureza da população de células cancerígenas.
Para data, muito poucos genes associados com subtipos de GC foram identificados. O CDH1
gene, que codifica a proteína E-caderina, os genes são os mais conhecidos associados com DGC hereditária (HDGC) [13-16]. rastreio genético para estas mutações tem sido sugerido, a fim de diagnosticar precoce GC [17]. disfunção de E-caderina, causada por mutações, perda de heterozigosidade, e hipermetilação do promotor, é o defeito mais bem estabelecida na iniciação e no desenvolvimento de GC [18-20]. Um estudo de associação do genoma mostrou que polimorfismos no gene do antigénio de células-tronco da próstata (PSCA
) estão fortemente associados com a susceptibilidade à DGC [21]. O método baseado em microarrays, no entanto, é limitada às variações de um único nucleótido, e não pode detectar variações estruturais cópia neutra (SVS). Dois estudos recentes relataram exomes GC, e mostrou que mutações no gene ARID1A
são frequentemente detectados em GC com instabilidade de microssatélites, e no vírus de Epstein-Barr (EBV) -positivo GCs [22, 23]. Nenhuma análise de subtipos GC foi realizada, ea maioria das amostras analisadas nos estudos eram de pacientes com IGC.
Next-Generation Sequencing (NGS) tem permitido aos pesquisadores detectar variações associados à doença, e ajudou a desvendar os mecanismos subjacentes do desenvolvimento da doença. Em particular, a sequenciação do genoma inteiro (WGS) pode detectar a maioria das variações genômicas, incluindo SVs, como intracromossómica e rearranjos intercromossômicas. Alternativamente, toda exome sequenciação (SIE), um método de sequenciação-alvo capturado, pode ser utilizado para a sequenciação de alta profundidade de um grande número de amostras a um custo relativamente baixo [24], embora apenas variações individuais de nucleótidos (SNVS) e pequenas inserções ou deleções (indels) podem ser identificados usando este método. WGS e WES cada um tem vantagens e desvantagens, e uma série de estudos recentes têm utilizado os dois métodos [25-27].
Aqui nós apresentamos a caracterização detalhada de genomas DGC de tumor encontrado e amostras normais, gerando perfis inteiros genômicas seguido por WES . Utilizou-se amostras de sangue como um controle normal, como em estudos anteriores [28-31]. A fim de encontrar variações DGC-específica, genomas IGC também foram analisadas e comparadas com as variações identificados no genoma de PED. A análise da estrutura de proteínas tridimensional foi realizada para novas mutações somáticas do gene da CDH1
, e este identificado regiões críticas que foram funcionalmente alteradas pelas mutações. Além disso, encontramos um gene de fusão romance que poderiam estar envolvidos na tumorigênese.
Resultados e discussão
genoma inteiro e sequenciamento exome
tumor e amostras pareadas normais (sangue) de 14 pacientes com DGC (as características clinicopatológicas desses pacientes são apresentados na Tabela S1 no arquivo adicionais 1), que eram todos relativamente jovens (idade média 38 anos) mulheres coreanas, foram sequenciados usando um Illumina HiSeq 2000, que produziu emparelhado-end, 90-base e 101 de base de DNA lê. Além disso, cinco pares de tumor e amostras normais pareados de pacientes com IGC (idade média de 42 anos) foram submetidos a sequenciação de ADN; uma destas amostras foi identificada mais tarde como um caso de instabilidade microssatélite (MSI) e, portanto, foi excluído da análise de mutações. Nenhuma das amostras teve qualquer história familiar de câncer, e os subtipos foram histologicamente confirmados. Apenas as células tumorais foram recolhidos por macrodissection após coloração hematoxilina. Compra de toda a análise do genoma, em média, 92 gigabases (GB) por amostra foram produzidos em cerca de 32 vezes a profundidade seqüenciamento, chegando a 3,5 terabases (TB) no total, e foram mapeado para o genoma de referência (NCBI construir 37, hg19) a uma velocidade de mapeamento maior do que 94,5% (para as estatísticas de sequenciação, ver arquivo adicional 1: Tabela S2). Usando a final 3,3 TB de mapeada lê, um banco de dados perfil genômico foi construído para SNVS detecção, copie variações no número (CNVs), e VV. Porque a pureza celular de uma amostra de tumor é um elemento fundamental para a análise do genoma do câncer, foi avaliada utilizando um método de cálculo em casa (ver Materiais e Métodos; ver arquivo adicionais 1: Tabela S3 e Figura S1). Embora nós tentamos coletar células tumorais apenas, nossas amostras ainda mostrou um alto nível de mistura do estroma. Para aumentar a precisão da detecção de mutações em regiões gênicas, mesmo em amostras de baixa pureza, WES adicional foi realizada a cerca de 103 vezes a profundidade sequenciamento em média, o que produziu um total de 17 dados de sequência GB. O WES capturado coberto 93,1% da região génica em 10 vezes ou maior profundidade, e esta cobertura é semelhante ao de dados exome previamente relatados em CG [22, 23].
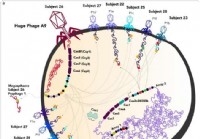
Combinando WGS os dados e Wes, detectou-somática alterações nas amostras de DGC e comparou-os com as alterações IGC (os dados estão resumidos na Figura 1 como um diagrama de circo). Para verificar nossos dados, combinadas e analisadas los com dados relatados anteriormente exome de dois estudos diferentes (24 CIG e 5 amostras DGC, não incluindo MSI e amostras mistas) [22, 23] e dados de hibridização matriz genoma comparativa (CGH) ( 16 CIG e 14 amostras DGC) [32]. Embora estes estudos utilizaram principalmente CIG e incluiu apenas um pequeno número de amostras DGC, eles poderiam ser complementares aos nossos dados como um controlo (por fornecimento de um aumento do número de dados e de eliminação da especificidade do tecido IGC). No conjunto de dados combinados, foram comparadas as diferenças de alterações entre as amostras DGC e IGC. Figura 1 Distribuição inteiro genoma de mutações somáticas e eventos de duplicação ou exclusão em cancros gástricos de tipo difuso (PED). Todas as mutações somáticas, incluindo eventos de duplicação /deleção, que foram encontrados nos 14 genomas DGC, são mesclados na trama circo. De fora para dentro, a trama apresenta as seguintes características: ideogramas cromossômicas, frequência de eventos cumulativos de amplificação ou supressão (preto, amplificação; vermelhos, eliminação), e o número de variações de nucleotídeo único não-sinônimas somáticas (nsSNVs), indels e SNVS em locais de junção para cada gene. triângulos pretos indicam genes altamente mutados. triângulos laranja indicam oncogenes e triângulos azuis indicam os supressores de tumor.
Identificação do SNVS difusa de tipo específico-e indels Online em cada par de amostra, foram identificados cerca de 3,7 milhões de SNVS, que foram verificados usando polimorfismo de nucleotídeo único (SNP ) chips (taxa média de concordância: 99,2%; ver arquivo adicionais 1: Tabela S4), e aproximadamente 0.690.000 indels (para detalhes, veja arquivo adicionais 1: Tabela S5 e Tabela S6). O primeiro avaliou a frequência mutacional dos dois tipos de GC no nível de nucleotídeo único (ver arquivo adicionais 1: Figura S2 a, b). O espectro de mutações somáticas foram dominados por C > T (G > A) em ambas as transições as amostras de DGC e IGC, e não houve diferenças significativas em contextos mutacionais entre os dois tipos de GC, de acordo com estudos anteriores de GC [23, 30]. Quando analisamos dois conjuntos de dados exome reportados anteriormente, verificou-se que o espectro da relação de substituição de nucleotídeos foi semelhante aos nossos dados (ver arquivo adicionais 1: Figura S2c, d).
Embora o espectro de mutações de DGC é semelhante ao de IGC, mutações individuais em genes afetados eram diferentes. Subtraindo mutações encontradas nos genomas de sangue normais, foram identificados 922 SNVS não sinónimas (nsSNVs) como mutações somáticas nas amostras tumorais 18 (ver arquivo adicionais 1: Tabela S7, ver arquivo adicionais 2). A taxa média de mutação dos 18 GCs (1,97 mutações /Mb) foi comparável com o relatado em outros estudos sobre cólon, pâncreas e câncer de fígado [33-35]. De 847 genes mutantes afetados pelos 922 nsSNVs, 581 foram em 14 casos DGC, 288 foram em 4 casos IGC, e 22 (2,6%) eram comuns a ambos os tipos. A amostra MSI, que foi excluído da análise comparativa, mostrou aproximadamente seis vezes mais SNVS e indels do que os outros amostras; este resultado está de acordo com um relatório anterior [22]. Quando combinados os dois conjuntos de dados exome previamente relatados, foram identificados 967 e 2.077 nsSNVs somáticas em 19 PED e 28 CIG, respectivamente. A taxa de mutação somática do CIG (3,71 mutações /Mb nas 28 amostras) foi maior do que a dos PED (2,29 mutações /Mb nas 19 amostras) (ver arquivo adicionais 1: Tabela S8). pesquisa publicado anteriormente sugere que o melanoma e cancro do pulmão têm altas taxas de mutação, devido ao envolvimento de agentes mutagénicos potentes [36]. Da mesma forma, é possível que IGC tem essa alta taxa de mutação porque seu mecanismo tumorigênico pode estar associado mais com meio ambiente e /ou mutagénicos parasitárias comparação com DGC. Compra de variações individuais, os genes do câncer causadores putativos foram previstos pelo motorista de cálculo da pontuação gene (ver arquivo adicionais 1: Tabela 1 e Tabela S9). O gene CDH1
foi encontrado para ser abundantemente mutado em DGC (P
= 1,29 × 10
-2), incluindo seis mutações somáticas (três missense, um absurdo, um frameshift e um mutações no local emenda) que não tenham sido previamente relatado, ao passo que apenas uma mutação sem sentido foi encontrado nas amostras IGC (Tabela 2). Todos os sete CDH1
mutações somáticas foram verificadas por seqüenciamento Sanger (ver arquivo adicionais 1: Tabela S10 e S11 Tabela). Nas nossas amostras DGC, 35,7% (5/14) teve CDH1
mutações somáticas, e foi relatado que as frequências de CDH1
mutações somáticas no PED esporádicos pode variar entre 3% e superior a 50% [19 , 37-40]. Verificou-se que em países com uma elevada incidência de CG esporádicos (como Japão e Coreia), a frequência de mutações germinativas em GCs familiar é baixa em comparação com a de países de baixa incidência [41, 42]. Portanto, especula-se que a incidência global de GC também está relacionado com a frequência de CDH1
mutações somáticas. Além disso, uma mutação germinativa (T340A) em CDH1
foi encontrado em ambos os tumores e os correspondentes genomas de sangue de duas amostras (D-14, DGC; M-01, MSI-tipo). Embora T340A é uma mutação causal na HDGC [43], estes dois pacientes não têm qualquer história familiar, como GC ou cancro da mama lobular. Dois relatórios anteriores análise de dados exome de GC não identificou CDH1
como um gene altamente classificado (apenas uma mutação sem sentido em uma amostra MSI IGC) [22, 23]. Esta discrepância pode ser devido ao pequeno número de amostras de DGC nesses estudos (2 de 22 e três das 15 amostras foram PED, respectivamente). No presente trabalho, PIK3CA Comprar e TP53
, conhecida genes associados ao câncer, eram os genes mais frequentemente mutado em ambos DGC e IGC ver Tabela 1 e Tabela S9 em arquivo adicionais 1. As mutações em dois PIK3CA conhecido
hotspots (E545K e H1047L) foram encontrados em quatro amostras DGC. Além disso, uma mutação nsSNV (Q546K) adjacente à mutação E545K foi encontrado em uma amostra de DGC. No total, 5 de 14 amostras de DGC (aproximadamente 30%), continham nsSNVs em PIK3CA
, que é um oncogene cuja mutadas exposições forma um aumento da actividade de cinase, provocando proliferação de células cancerosas [44]. Nós, então, comparada a baixa frequência (16-17%) dos nsSNVs em PIK3CA
em relatórios por outros [22, 23, 44] (que amostras IGC usado principalmente) e os resultados de nossa análise combinada (31,5% para DGC , 14,3% para a CIG) (ver arquivo adicionais 1: Tabela S9). Afigura-se que as taxas relativamente elevadas de mutação de PIK3CA
em DGC pode reflectir a especificidade das mutações neste gene para este tipo de cancro. Além disso, três amostras (dois DGC e uma CIG) continha tanto nsSNV e uma perda de cópia de TP53
, indicando uma perda de função em homozigotos
TP53, tal como anteriormente relatado [45]. Um SNP no gene de PSCA
(rs2976329) foi avaliado para ser associada com um risco aumentado de DGC em populações japonesas e coreanas [21]. Este SNP também foi enriquecido na maioria das amostras de DGC em nosso estudo, (9 em cada 14 pacientes), indicando que nossas amostras analisadas representam pacientes típicos com DGC na Ásia Oriental. Além disso, uma mutação nonsense (R1446 *) na ARID1A
gene, foi encontrado em uma amostra DGC (D-08). Embora mutações no ARID1A
são frequentemente detectados no MSI e, em GCs EBV-positivos [22, 23], a amostra D-08 não apresentaram infecção por EBV, e uma amostra MSI (M-01) não tem qualquer ARID1A
mutações genéticas tampouco. De variações de genes motorista candidatos, 88 nsSNVs, 4 pequenas Indels, e 2 SNVS em um local de junção foram verificadas utilizando sequenciamento Sanger convencional. Sete destas mutações não poderia ser testada por causa da falha PCR, e dos restantes 87 mutações, 96,6% foram confirmados como verdadeiros mutações somáticas (ver arquivo adicionais 1: Tabela S10 e S11 Tabela) .table 1 Principais genes motorista candidatos em 14 difusa cancros do tipo gástrico
Gene
Samples, n
nsSNVs, n
SNVS em local de união, n
Indels, n
P
-valor
driver pontuação gene PIK3CA
5
5
0
0
3,63 × 10 -12
9,83
CDH1
5 4
1
 O café ajuda a desenvolver micróbios intestinais saudáveis e evita os movimentos intestinais
O café ajuda a desenvolver micróbios intestinais saudáveis e evita os movimentos intestinais
 Grandes fagos recém-descobertos borram a fronteira entre vida / não vida
Grandes fagos recém-descobertos borram a fronteira entre vida / não vida
 Micróbios intestinais podem estar ligados à depressão
Micróbios intestinais podem estar ligados à depressão
 Microbioma de espermatozoides revelado com sequenciamento de RNA
Microbioma de espermatozoides revelado com sequenciamento de RNA
 Perfectus Biomed apresentará na conferência IPS em Liverpool
Perfectus Biomed apresentará na conferência IPS em Liverpool
 Uma abordagem multimônica para o desenvolvimento de medicamentos contra COVID-19
Uma abordagem multimônica para o desenvolvimento de medicamentos contra COVID-19
 Bactéria intestinal mais saudável com dieta à base de plantas ou mediterrânea
Um novo estudo mostra que alimentos específicos fornecidos por uma dieta à base de vegetais ou do tipo mediterrânea podem proteger o intestino contra doenças inflamatórias, promovendo seletivamente o
Bactéria intestinal mais saudável com dieta à base de plantas ou mediterrânea
Um novo estudo mostra que alimentos específicos fornecidos por uma dieta à base de vegetais ou do tipo mediterrânea podem proteger o intestino contra doenças inflamatórias, promovendo seletivamente o
 A pesquisa analisa os aerobiomas,
árvores e implicações para a saúde pública Os microrganismos ambientais desempenham um papel essencial na saúde humana - quanto diverso o consórcio, o melhor. A diversidade de microorganismos ajuda o
A pesquisa analisa os aerobiomas,
árvores e implicações para a saúde pública Os microrganismos ambientais desempenham um papel essencial na saúde humana - quanto diverso o consórcio, o melhor. A diversidade de microorganismos ajuda o
 A terapia biológica pode diminuir o risco de COVID-19 grave
Um estudo observacional recente de pesquisadores espanhóis, atualmente disponível no medRxiv * servidor de pré-impressão, sugere que os pacientes com doenças imunomediadas que recebem terapia biológ
A terapia biológica pode diminuir o risco de COVID-19 grave
Um estudo observacional recente de pesquisadores espanhóis, atualmente disponível no medRxiv * servidor de pré-impressão, sugere que os pacientes com doenças imunomediadas que recebem terapia biológ