oppdage menneskets historie fra magebakterier
Abstract
Nye analyser av menneskelige patogener har avdekket at deres evolusjonære historie er sammenfallende med den hypotese mønster av gamle og moderne menneskelige befolkning vandringer. Fylogenetiske trær av stammer av bakterien Helicobacter pylori Hotell og polyoma JC-viruset tatt fra geografisk forskjellige grupper av mennesker korrelerer tett med relasjoner av befolkningen der de finnes.
Charles Darwin anerkjent at fordelingen og form parasitter var evolusjonært viktig. Han bemerket for eksempel at "... det Pediculi [lus] samlet inn i ulike land fra de ulike raser av mennesker ... forskjellig, ikke bare i farger, men i strukturen av klørne og lemmer. I hvert fall i hvilke mange prøver ble oppnådd forskjellene var konstant "[1]. Mer nylig har flere forskningsgrupper [2-7] funnet interessante sammenhenger mellom de evolusjonære relasjonene mellom ulike bakterie- og virusstammer hostet av mennesker og mønsteret av vandringer av moderne mennesker over hele verden., En spesielt interessant sak er at av Helicobacter pylori
, en Gram-negative bakterien forbundet med gastritt, magesår og magekreft som kan infisere inntil halvparten av alle mennesker [8]. Oppdagelsen av at en bakteriell infeksjon kan føre til det ble ansett kroniske sykdommer [8] var et slående eksempel på at smittsomme sykdommer ennå ikke er erobret. Den fortsatte ervervet immunsvikt syndrom (AIDS) epidemien utbrudd av Ebola i Sentral-Afrika, og dagens spredning av West Nile Virus i USA og av Sars (SARS) fra Asia dokumentere pervasiveness og helsemessige konsekvenser av smittestoffer selv i en alder av vaksinering og antimikrobiell og antiviral terapi. Mange infeksjonssykdommer er antatt å ha oppstått samtidig med utviklingen av jordbruket og fremveksten av urban livsstil. Hvis du istedet mange patogener 'relasjoner med mennesker er mye eldre, ville det ikke være overraskende å finne dypere evolusjonære sammenhenger mellom mennesker og deres mikrobiell og viral inntrengere.
Evolusjonære historien til H. pylori
kan gi et eksempel av koevolusjon av en bakterie, og dens eneste kjente vert. Den H. pylori
genom er relativt små på 1,67 megabases, med en minimal bemanning av metabolske gener [9]. Variasjon mellom H. pylori
isolater fra forskjellige personer eller fra en person er stor, noe som fører til unike fingeravtrykk for nesten hvert isolat så langt skrevet. De kodende genene ikke er svært forskjellige, men: det meste av variasjonen skjer i den tredje base posisjon innenfor kodoner eller gjennom inversjoner eller translo-kationer, slik at de kodede aminosyre-sekvensene relativt lik [2]. Denne aminosyre-sekvenskonservering er heldig for vaksine forskning, som en vaksine er sannsynlig å være effektive på mange stammer. Mer interessant er det faktum at H. pylori
har en ekstremt høy grad av rekombinasjon, høyere faktisk enn for noen annen organisme, karakterisert este [3, 10].
Normalt en så høy hastighet av rekombinasjon ville gjøre dedusere en organismes evolusjonære historie svært vanskelig, ettersom informasjon om opprinnelsen til hver mutasjon ville gå tapt som det sprer seg i hele befolkningen. Men kombinert med modus for overføring av H. pylori
, kan denne ekstremt høye frekvensen av rekombinasjon faktisk gjøre evolusjonære slutninger enklere. Flere studier tyder sterkt på at H. pylori
er vanligvis sendes innen familier, vanligvis fra mor til barn [11, 12]. Dermed overføring av H. pylori
på noen måter ligner at av matern overføres mitokondrie-DNA [13]. Fordi mitokondrie DNA overføres utelukkende fra en av foreldrene (mor), og ikke rekombinere, har det vist seg å være en ideell genetisk system for å utlede human utviklingshistorie (se nedenfor) [14, 15]. Hvis H. pylori
er faktisk overveiende maternale overføres, vil nye stammer vanligvis ikke infisere en person i løpet av livet; sammen med den høye frekvensen av rekombinasjon, vil dette bety at de mutasjoner som akkumuleres i bestanden av bakterier i en persons mage vil være relativt homogen. Dette skal resultere i en sverm av stammer som er svært nært knyttet til hverandre, som inneholder mange av de mutasjoner som har skjedd i de enkelte bakterier. Svermer finnes i ulike mennesker vil dermed være mer forskjellige fra hverandre enn om det var mindre rekombinasjon.
Fleste smittsomme sykdommer spredte seg raskt over hele verden og stammer fra ulike regioner er relativt like, men førstegangskontroll av H. pylori
fra folk fra ulike deler av verden avslørt ganske sterk geografisk fordeling i europeiske og asiatiske H. pylori
typer [2-4]. Nylig, Falush og medarbeidere [5] har undersøkt dette partisjonering i større detalj. Etter sekvense åtte gener - totalt 3.850 nukleotider - i 370 stammer avledet fra 27 befolkningsgrupper, fant de 1,418 polymorfe nucleotide posisjoner. Deretter påføres et nytt analytisk verktøy, struktur [16], som ble utviklet for å utlede menneskets genetiske struktur fra multilocus genotype data. Programmet bruker Bayesianske metoder for å identifisere subgrupper med karakteristiske allelfrekvenser, og klynger av undergruppene, selv i nærvær av rekombinasjon [16]. Når denne teknikken ble brukt til H. pylori
sekvenser, ble fire hoved klynger funnet - to fra Afrika og en hver fra Europa og Asia (figur 1a) [5]. Figur 1 Forholdet mellom befolkningsgrupper, som beregnet fra H. pylori
funnet i magen og fra mitokondrielle DNA-data. (A) Forholdet mellom moderne subpopulasjoner av H. pylori product: [5]. Hver undergruppe er representert ved en sirkel med en diameter proporsjonal med det genetiske mangfoldet i den. Sentrene av sirklene er sammen med en fylogenetisk tre som viser forholdet mellom de fire undergruppene. Bakterier i hver undergruppe finnes hovedsakelig hos personer som stammer fra regionene vises. (B) En populasjonsnivå fylogenetisk tre av H. pylori
geografiske subpopulasjoner vist i (a). (C) En median-sammenføyning nettverk av menneskelige populasjoner avledet fra mitokondrie-DNA [14]. Et slikt nettverk viser alternative mulige evolusjonære relasjonene mellom klynger. Hver sirkel representerer en klynge av mitokondrielle typer med en diameter som er proporsjonal med frekvensen av den typen innenfor undergruppene. Alle ikke-afrikanske populasjoner er utledet fra en afrikansk avstamning; nettverket av relasjoner innen denne linjen er forstørret (øverst). (A, b) Hentet fra [5]; (C) innrettet til fra [14]
Hver klynge funnet ved Falush og medarbeidere [5] kan bli delt inn i undergrupper.; for eksempel, kan den "Africa 1 'cluster deles videre inn i Vesten og sørafrikanske subclusters, og Øst-Asia klyngen kan bli delt i østasiatisk, Amerind, og Maori subclusters. Den geografiske fordeling innenfor de 200 europeiske stammene var spesielt komplisert, antagelig fordi mange grupper har blåst frem og tilbake over hele Europa i løpet av de siste flere årtusener. Europeiske stammer også fra tid til annen dukket opp i Amerika, Australia, og blant sørafrikanere, antagelig reflekterer kolonierobringer.
Den fylogenetiske relasjoner av disse klyngene (Figur 1b) og deres underavdelinger [5] viser et mønster som ligner den som oppnås ved hjelp av mitokondrie-DNA variant (figur 1c) [14, 15]. Det moderne menneskets gener, som utledes fra mitokondrie-DNA og bekreftet av Y kromosom studier, er antatt å ha hatt en afrikansk opprinnelse ca 150,000-200,000 år siden [14, 15, 17, 18]. Den opprinnelige befolkningen så spredt og variert hele Afrika for nesten 100.000 år, før ekspandere inn i Vest-Asia og Europa og i Sør- og Øst-Asia ca 50.000-60.000 år siden, og erstatte de eksisterende arkaiske bestander av mennesker i disse regionene. Påfølgende vandringer spredt i Australasia av 40.000 år siden, deretter til øyene i Stillehavet, og senere i Nord-Amerika, ca 15 000 år siden (figur 2) [14, 15, 17, 18]. Den bemerkelsesverdige likheter mellom dette synet på menneskets historie og resultater fra studier av H. pylori
har ført Falush og kolleger [5] samt andre [2] til å konkludere med at H. pylori
evolusjonen har fulgt stien av moderne menneskelig ekspansjon og migrasjon. Dette arbeidet gir dermed en annen type data for analyse av menneskelig evolusjon og migrasjon, uavhengig av mitokondriell DNA og Y-kromosomer, som vil være verdifulle i videre studier. Dessverre, men anslå avvik datoer fra H. pylori
er spesielt vanskelig, på grunn av den ekstremt høye frekvensen av rekombinasjon [10]. Ytterligere prøvetaking og analyseteknikker kan være nødvendig å ytterligere teste den vandrende hypotesen. Figur 2 Et kart av mønsteret av ekspansjon og migrering av moderne mennesker over hele verden, avledet fra studier av mitokondrisk DNA og Y-kromosomer [14, 15, 17, 18]. Tallene angir omtrentlig tid (i år før i dag) da det moderne mennesket først dukket opp i den angitte regionen.
Andre patogener har også vært foreslått å følge evolusjonære historier som ligner på sine verter. Et av de mest interessante eksempler, men med en ikke-human vert, er det av bladlus, en bakterie som finnes i dem, og to plasmider er forbundet med bakterien. Funk et al.
[19] fant at de utledede intraspecifrc phylogenies av disse fire genomer var helt sammenfallende. Retur til menneskelige patogener, kan den menneskelige polyomavirus JC virus (JCV) deles inn i genotyper som svarer til de store kontinentale landmassene [6]. Som H. pylori
, JCV - noe som kan forårsake progressiv multifokal leukoencefalopati (tap av myelinisering i sentralnervesystemet) - er meget utbredt blant mennesker som et resultat av familiær overføring. Totalt 12 kjente undergrupper er definert, med europeiske, afrikanske og asiatiske distribusjoner [7]. Selv om direkte slutninger av en afrikansk opprinnelse for JCV er problematisk fordi det ikke er egnet outgroup med å heie fylogenetisk tre, når en afrikansk opprinnelse antas en rimelig evolusjonære historie kan bli en hypotese (figur 3) [7]. Som med H. pylori
, dedusere molekylær divergens datoer er for tiden problematisk for JCV. Videre etterforskning i den evolusjonære historien til disse menneskelige patogener er derfor nødvendig. Figur 3 Relasjoner av menneskelig polyoma JC virus (JCV) undertyper som finnes i mennesker fra ulike deler av verden [7]. Bokstavene referer til individuelle undergrupper. (A) en hypotese mønster av spredning av JCV subtyper gjennom verden (unntatt Amerika); (B) en antatt fylogeni av JCV subtyper, forutsatt en afrikansk opprinnelse for viruset. Tilpasset fra [7].
Klargjørende mønstre av utviklingen av menneskelige patogener kan til slutt gi ytterligere bevis ikke bare om deres historie, men også om menneskets evolusjon og historie. Dette vil være spesielt for patogener som H. pylori
som har en overveiende mor-barn-modus for overføring, mimicking mitokondrie-DNA evolusjon. H. pylori
er utløsende rolle i flere kroniske mage forhold er bare ett av mange aktuelle eksempler på kjent eller mistenkt rollen som en smittestoff som fører til kroniske sykdommer. Bakterier er mistenkt for å være involvert i utviklingen av arteriosklerose, slag, og Crohns sykdom, mens virus er kjent for å føre til AIDS og de forskjellige former for kronisk hepatitt. Livmorhalskreft, leverkreft, er Burkitt lymfom, Kaposis sarkom, og kanskje diabetes mellitus også enten kjent eller mistenkt for å være av viral opprinnelse. Dersom noen av de allestedsnærværende kroniske sykdommer vise seg å være av bakteriell eller viral opprinnelse, bør hele verden undersøkelser av disse patogener i humane populasjoner utføres umiddelbart, slik at kunnskap om utviklingen og mangfoldet av disse patogener kan bli innarbeidet i forskningsinnsatsen som skal lindre forholdene som de forårsaker.
 Glycyrrhizic acid som stoffkandidat for COVID-19
Glycyrrhizic acid som stoffkandidat for COVID-19
 Rotavirus spiller en rolle i utviklingen av type 1 diabetes
Rotavirus spiller en rolle i utviklingen av type 1 diabetes
 Forskere håper blodprøver som nøyaktig diagnostiserer fibromyalgi kan være tilgjengelig innen fem år
Forskere håper blodprøver som nøyaktig diagnostiserer fibromyalgi kan være tilgjengelig innen fem år
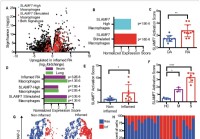 Ny superaktiverende makrofagreseptor kan forklare hyperinflammasjon ved alvorlig COVID-19
Ny superaktiverende makrofagreseptor kan forklare hyperinflammasjon ved alvorlig COVID-19
 Sædmikrobiom avslørt med RNA -sekvensering
Sædmikrobiom avslørt med RNA -sekvensering
 Kan antivirale avledninger av mikroalger bekjempe SARS-CoV-2 og andre virus?
Kan antivirale avledninger av mikroalger bekjempe SARS-CoV-2 og andre virus?
 Ulcerøs kolitt og en manglende mikrobe i tarmen
Ulcerøs kolitt er en alvorlig svekkende inflammatorisk sykdom i tarmen som fører til lammende symptomer som kan påvirke livskvaliteten alvorlig. Forskere fra Stanford University School of Medicine har
Ulcerøs kolitt og en manglende mikrobe i tarmen
Ulcerøs kolitt er en alvorlig svekkende inflammatorisk sykdom i tarmen som fører til lammende symptomer som kan påvirke livskvaliteten alvorlig. Forskere fra Stanford University School of Medicine har
 COVID-19:Hvordan sykdommen ser ut
Med økende informasjon om COVID-19 sykdom samlet førstehånds, et mer nøyaktig bilde av de kliniske trekkene kommer sammen. Hvordan COVID-19 presenterer seg Så hvordan starter det? Statlige helsede
COVID-19:Hvordan sykdommen ser ut
Med økende informasjon om COVID-19 sykdom samlet førstehånds, et mer nøyaktig bilde av de kliniske trekkene kommer sammen. Hvordan COVID-19 presenterer seg Så hvordan starter det? Statlige helsede
 Sunneste tarmbakterier med plantebasert eller middelhavskosthold
En ny studie viser at spesifikke matvarer som er levert av enten en plantebasert eller en middelhavskost, kan beskytte tarmen mot inflammatoriske lidelser, ved selektivt å fremme veksten av antiinflam
Sunneste tarmbakterier med plantebasert eller middelhavskosthold
En ny studie viser at spesifikke matvarer som er levert av enten en plantebasert eller en middelhavskost, kan beskytte tarmen mot inflammatoriske lidelser, ved selektivt å fremme veksten av antiinflam