El nuevo método identifica grupos de bacterias ecológica y médicamente relevantes.
La identificación de especies entre plantas y animales ha sido una ocupación de tiempo completo para algunos biólogos, pero la tarea es aún más abrumadora para la miríada de microbios que habitan el planeta. Ahora, Los investigadores del MIT han desarrollado una medida simple del flujo de genes que puede definir poblaciones ecológicamente importantes entre bacterias y arqueas. incluida la localización de poblaciones asociadas con enfermedades humanas.
La métrica de flujo de genes separa los microbios coexistentes en poblaciones genéticamente y ecológicamente distintas, Martín Polz, profesor de ingeniería civil y ambiental en el MIT, y colegas escriben en la edición del 8 de agosto de
Celda. Polz y sus colegas también desarrollaron un método para identificar partes del genoma en estas poblaciones que muestran diferentes adaptaciones que pueden mapearse en diferentes entornos. Cuando probaron su enfoque en una bacteria intestinal, por ejemplo, pudieron determinar que diferentes poblaciones de bacterias estaban asociadas con individuos sanos y pacientes con enfermedad de Crohn.
Los biólogos a menudo denominan especie a un grupo de plantas o animales si el grupo está aislado reproductivamente de los demás, es decir, los individuos del grupo pueden reproducirse entre sí, pero no pueden reproducirse con otros. Como resultado, los miembros de una especie comparten un conjunto de genes que se diferencia de otras especies. Gran parte de la teoría de la evolución se centra en especies y poblaciones, los representantes de una especie en un área particular.
Pero los microbios "desafían el concepto clásico de especies de plantas y animales, "Explica Polz. Los microbios tienden a reproducirse asexualmente, simplemente dividiéndose en dos en lugar de combinar sus genes con otros individuos para producir descendencia. Los microbios también son conocidos por "tomar ADN de fuentes ambientales, como virus, ", dice." Los virus pueden transferir ADN a células microbianas y ese ADN puede incorporarse a sus genomas ".
Estos procesos dificultan la clasificación de los microbios coexistentes en poblaciones distintas en función de su composición genética. "Si no podemos identificar esas poblaciones en microbios, no podemos aplicar uno a uno toda esta rica teoría ecológica y evolutiva que se ha desarrollado para plantas y animales a los microbios, "dice Polz.
Si los investigadores quieren medir la resiliencia de un ecosistema frente al cambio ambiental, por ejemplo, podrían observar cómo cambian las poblaciones dentro de las especies a lo largo del tiempo. "Si no sabemos qué es una especie, es muy difícil medir y evaluar este tipo de perturbaciones, " él añade.
Un criterio para el flujo de genes Martin y sus colegas decidieron buscar otra forma de definir poblaciones de microbios ecológicamente significativas. Dirigido por el estudiante graduado de microbiología Philip Arevalo, Los investigadores desarrollaron una métrica de flujo de genes que llamaron PopCOGenT (Poblaciones como clústeres de transferencia de genes).
PopCOGenT mide el flujo de genes reciente o la transferencia de genes entre genomas estrechamente relacionados. En general, Los genomas microbianos que han intercambiado ADN recientemente deberían compartir tramos más largos y frecuentes de ADN idéntico que si los individuos se estuvieran reproduciendo dividiendo su ADN en dos. Sin este tipo de intercambio reciente, los investigadores sugirieron, la longitud de estos tramos compartidos de ADN idéntico se acortaría a medida que las mutaciones insertaran nuevas "letras" en el tramo.
Dos cepas microbianas que no son genéticamente idénticas entre sí pero que comparten "trozos" considerables de ADN idéntico probablemente estén intercambiando más material genético entre sí que con otras cepas. Esta medición del flujo de genes puede definir distintas poblaciones microbianas, como descubrieron los investigadores en sus pruebas de tres tipos diferentes de bacterias.
En
Vibrio bacterias por ejemplo, poblaciones estrechamente relacionadas pueden compartir algunas secuencias de genes centrales, pero parecen completamente aislados entre sí cuando se ven a través de esta medición del flujo genético reciente, Polz y sus colegas encontraron.
Polz dice que el método PopCOGenT puede funcionar mejor para definir poblaciones microbianas que los estudios anteriores porque se centra en el flujo de genes reciente entre organismos estrechamente relacionados. en lugar de incluir eventos de flujo de genes que pueden haber ocurrido hace miles de años.
El método también sugiere que, si bien los microbios están absorbiendo constantemente ADN diferente de su entorno, lo que podría oscurecer los patrones de flujo de genes, "puede ser que este ADN divergente se elimine realmente mediante la selección de poblaciones muy rápidamente, "dice Polz.
El enfoque de la ecología inversa El estudiante graduado de microbiología David VanInsberghe sugirió entonces un enfoque de "ecología inversa" que podría identificar regiones del genoma en estas poblaciones recién definidas que muestran "barridos selectivos", lugares donde la variación del ADN se reduce o se elimina. probablemente como resultado de una fuerte selección natural de una variante genética beneficiosa en particular.
Al identificar barridos específicos dentro de las poblaciones, y mapear la distribución de estas poblaciones, el método puede revelar posibles adaptaciones que impulsan a los microbios a habitar un entorno o un huésped en particular, sin ningún conocimiento previo de su entorno. Cuando los investigadores probaron este enfoque en la bacteria intestinal
Ruminococcus gnavus , descubrieron poblaciones separadas del microbio asociado con personas sanas y pacientes con enfermedad de Crohn.
Polz dice que es probable que el método de ecología inversa se aplique en un futuro próximo para estudiar la diversidad completa de las bacterias que habitan el cuerpo humano. "Hay mucho interés en secuenciar organismos estrechamente relacionados dentro del microbioma humano y buscar asociaciones de salud y enfermedad, y los conjuntos de datos están creciendo ".
Espera utilizar el enfoque para examinar el "genoma flexible" de los microbios. Cepas de
E. coli bacterias por ejemplo, comparten alrededor del 40 por ciento de sus genes en un "genoma central, "mientras que el otro 60 por ciento, la parte flexible, varía entre las cepas". Para mí, es una de las preguntas más importantes en microbiología:¿Por qué estos genomas son tan diversos en contenido genético? ”, explica Polz.“ Una vez que podamos definir las poblaciones como unidades evolutivas, podemos interpretar las frecuencias de genes en estas poblaciones a la luz de los procesos evolutivos ".
Los hallazgos de Polz y sus colegas podrían aumentar las estimaciones de la diversidad microbiana, dice Marx. "Lo que creo que es realmente genial acerca de este enfoque del grupo de Martin es que en realidad sugieren que la complejidad que vemos es incluso más compleja de lo que le damos crédito. Puede haber incluso más tipos que son ecológicamente importantes por ahí, cosas que si fueran plantas y animales los llamaríamos especies ".
Otros autores del MIT en el artículo incluyen a Joseph Elsherbini y Jeff Gore. La investigación fue apoyada, en parte, por la Fundación Nacional de Ciencias y la Fundación Simons.
 La tecnología de chips de órganos mejora el estudio del intestino para la medicina personalizada
La tecnología de chips de órganos mejora el estudio del intestino para la medicina personalizada
 Los probióticos pueden ofrecer beneficios terapéuticos para pacientes biopolares
Los probióticos pueden ofrecer beneficios terapéuticos para pacientes biopolares
 Una nueva estrategia puede fortalecer la comunicación entre el intestino y el cerebro
Una nueva estrategia puede fortalecer la comunicación entre el intestino y el cerebro
 Los pacientes con SII podrían beneficiarse de los suplementos de vitamina D,
Los pacientes con SII podrían beneficiarse de los suplementos de vitamina D,
 El trasplante de líquido vaginal podría ayudar a tratar la vaginosis bacteriana recurrente
El trasplante de líquido vaginal podría ayudar a tratar la vaginosis bacteriana recurrente
 Los microbiomas de primates antiguos pueden proporcionar más información sobre el desarrollo humano
Los microbiomas de primates antiguos pueden proporcionar más información sobre el desarrollo humano
 ¿El coronavirus se transmite a través de las heces?
Un sorprendente estudio de China, publicado en la revista Gastroenterología en marzo de 2020, informa que el nuevo coronavirus SARS-CoV-2 se puede propagar a través de las heces, así como a través d
¿El coronavirus se transmite a través de las heces?
Un sorprendente estudio de China, publicado en la revista Gastroenterología en marzo de 2020, informa que el nuevo coronavirus SARS-CoV-2 se puede propagar a través de las heces, así como a través d
 Los plásticos ahora se encuentran comúnmente en las heces humanas
Casi ocho mil millones de toneladas métricas de plástico llegan a los océanos cada año. Esta enorme cantidad de plástico se lava en tierra o se rompe en pequeños trozos de menos de 5 milímetros de diá
Los plásticos ahora se encuentran comúnmente en las heces humanas
Casi ocho mil millones de toneladas métricas de plástico llegan a los océanos cada año. Esta enorme cantidad de plástico se lava en tierra o se rompe en pequeños trozos de menos de 5 milímetros de diá
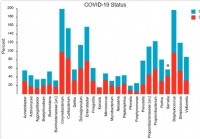 La composición y estructura del microbioma nasofaríngeo se relacionan con la gravedad de la enfermedad COVID-19
Las infecciones virales están asociadas con cambios en el microbioma del tracto respiratorio superior / nasofaríngeo (NP). Además, Muchos estudios plantean la hipótesis de las posibilidades de superin
La composición y estructura del microbioma nasofaríngeo se relacionan con la gravedad de la enfermedad COVID-19
Las infecciones virales están asociadas con cambios en el microbioma del tracto respiratorio superior / nasofaríngeo (NP). Además, Muchos estudios plantean la hipótesis de las posibilidades de superin