Extracto
El cáncer gástrico es una de las principales causas de muerte por cáncer en todo el mundo. Mientras que las formas hereditarias de cáncer gástrico son relativamente raros, la identificación de los genes responsables de estos casos puede informar el diagnóstico y el tratamiento para ambos casos hereditarios y esporádicos de cáncer gástrico. Las mutaciones en el gen de la E-cadherina, CDH1 Autor Resumen Las mutaciones genéticas subyacentes implicados en el 60% de los casos de cáncer gástrico heredados siguen siendo desconocidos. Aquí presentamos un gran pedigrí, ampliado con cáncer gástrico familiar y una asociación en parte de la familia con una mutación en MAP3K6 Visto:. Gastón D, Hansford S, C Oliveira, Nightingale M, H Pinheiro, MacGillivray C, et al. (2014) de la línea germinal mutaciones en MAP3K6 Editor: Marshall S. Horwitz, Universidad de Washington, Estados Unidos de América Recibido: March 3, 2014; Aceptado: August 14, 2014; Publicado: 23 de octubre 2014 Derechos de Autor © 2014 de Gaston et al. Este es un artículo de acceso abierto distribuido bajo los términos de la licencia Creative Commons Attribution License, que permite el uso ilimitado, distribución y reproducción en cualquier medio, siempre que el autor original y la fuente se acreditan Financiación:. La siguiente agencias proporcionado fondos para este proyecto: Genoma Canadá, Genoma Atlántico, Fundación para la Investigación de la Salud de Nueva Escocia, Nueva Escocia Investigación e Innovación Confianza, Dalhousie Facultad de Medicina, Departamento de Oftalmología de Dalhousie, Health Canada, el Centro de Investigación y Desarrollo de Medicamentos, Distrito capital Salud autoridad, IWK Health Foundation Centre, Fondo de Investigaciones Sanitarias de capital, y la Fundación COMPETE /FEDER portugués de Ciencia y Tecnología (FCT), Proyectos Ref. FCT PTDC ///79499/2011 de HP "financiados sin Ambito do Programa Operacional Temático Factores de Competitividade (competición) e comparticipado pelo fundo Comunitário Europeu FEDER." Es compatible SAU-GMG 110785/2009 y Post-doc SFRH subvención /TLP MES por el CHU Ste-Justine Centro de Investigación. Los autores desean reconocer la contribución de: la Plataforma de Secuenciación del Genoma Quebec alto rendimiento; y Sónia Sousa y José Carlos Machado de la Unidad de Diagnóstico IPATIMUP, Oporto, Portugal. Los donantes no tenía papel en el diseño del estudio, la recogida y análisis de datos, decisión a publicar, o la preparación del manuscrito Conflicto de intereses:.. Los autores han declarado que no existen intereses en competencia Introducción el cáncer gástrico es la segunda causa principal de muerte relacionada con el cáncer en todo el mundo, con 738.000 muertes por año [1]. El tratamiento primario consiste en la resección quirúrgica del tumor y puede ser seguida de quimioterapia y /o radioterapia. Las tasas de supervivencia a 5 años después de la resección quirúrgica son altos si la enfermedad se detecta a tiempo (71% para la etapa 1A), sin embargo, que haya que dejar fuera rápidamente cuando el diagnóstico se realiza en etapas posteriores (46% estadio IIA, 20% en estadio IIIA, 4% en estadio IV) (base de datos SEER del Instituto Nacional del cáncer, octubre 2013). Desafortunadamente, debido a los primeros síntomas de cáncer gástrico se parecen mucho a otras enfermedades, la detección a menudo no se produce hasta fases avanzadas ya se han llegado a [2] Clásicamente, cáncer gástrico ha sido dividido en dos tipos:. Intestinal y difuso [ ,,,0],3]. La forma intestinal se produce de forma espontánea y se encuentra más frecuentemente en personas de edad avanzada, mientras que la forma difusa se produce a menudo en los individuos más jóvenes y puede estar asociada con una historia familiar de cáncer gástrico. Las poblaciones con mayor prevalencia de Helicobacter pylori Hemos comprobado una gran familia de Canadá marítimo con una historia familiar de cáncer gástrico (FGC) presentan un patrón autosómico dominante aparente de la herencia, pero que no lleva variantes en la región de codificación de la CDH1 Evaluación clínica y patológica Hemos comprobado una gran marítimo canadiense familia de ascendencia europea en el transcurso de la evaluación clínica de rutina en la clínica de cáncer hereditario como parte de Marítima Médico Servicio de Genética del Centro de Salud IWK en Halifax, Nueva Escocia, Canadá (Figura 1). Saliva, sangre o muestras (FFPE) incluidas en parafina fijadas con formalina se obtuvieron a partir de 6 miembros de la familia con cáncer gástrico, así como 27 familiares no afectados, y un individuo casado-in. Sin consanguinidad era sospechoso en este pedigrí. El caso índice, afectado individuo de 1884, se le diagnosticó un carcinoma gástrico metastásico y se sometió a una gastrectomía total a la edad de 51. El examen anatomopatológico reveló un adenocarcinoma pobremente diferenciado que surge en el antro del estómago en un fondo de metaplasia intestinal y gastritis crónica. El tumor estaba compuesto de una lámina de células en anillo de sello (Figura 2C y D). El carcinoma penetrado a través de todo el espesor de la muscularis propia participación de la capa serosa. No hay evidencia de H. pylori tía materna del caso índice, individuo de 1826, se le diagnosticó carcinoma gástrico a los 80 años y se sometió a una gastrectomía parcial. El examen anatomopatológico reveló una moderada a adenocarcinoma pobremente diferenciado invadiendo en la muscular propia (Figura 2A). El tumor fue predominantemente compuesto de nidos cohesivos de células neoplásicas con formación glandular ocasional. se observaron células tumorales con las formas de células en anillo de sello en áreas sólidas (Figura 2B). La mucosa gástrica adyacente al tumor mostró metaplasia intestinal focal sin evidencia de H. pylori A los 76 una biopsia de estómago de otra tía materna de la persona afectada, la edad individuo 1841, se informó de que un adenocarcinoma moderadamente diferenciado con la formación glandular. H. pylori Una biopsia de estómago a los 82 años de paciente 1844, la madre del probando, mostró un adenocarcinoma pobremente diferenciado con características anillo de sello. La mucosa fondo mostró evidencia de H. pylori Paciente 1845, un primer primo una vez quitado para el probando, fue diagnosticado a los 59 años con un carcinoma indiferenciado y sin características del anillo de sello. El tumor se asoció con denso infiltrado linfoide y se clasificó mejor que el carcinoma linfoepitelial. H. pylori Tras la proyección de un panel de 115 probandos con CDH1 El cáncer gástrico se ha descrito para los pacientes 1845 y 2447 no tenía anillos de sello observaron y se le diagnosticó a una edad promedio anterior ( 52 frente a 72, aunque el individuo estudiado fue diagnosticado a los 51 años). Sobre la base de las diferencias en la histología, particularmente la falta de células en anillo de sello en 1845 y 2447 en comparación con los demás individuos afectados, es posible que la enfermedad en estos dos individuos representa una condición distinta. Alternativamente, es posible que la familia está mostrando un patrón fenotípico más compleja sea conducido por dos (o más) genes. A pesar de que el 30-40% CGDH de los casos son atribuibles a mutaciones en CDH1 También realizó vinculación no paramétrico (NPL), con un método un menor número de suposiciones subyacentes acerca del modelo de herencia subyacente, utilizando los individuos afectados por 1826, 1841, 1844 y 2447 (en todo el pedigrí) o con la eliminación de 2447 como un potencial phenocopy especificando que sean de estatus sanitario desconocido (sub-pedigrí). genómica intervalos de todo el pedigrí eran en su mayoría consistentes con los identificados utilizando los dos modelos paramétricos (Tabla 3). Exclusión de 2.447 dio lugar a una menor puntuación máxima total (1.204), que se encontró en varios intervalos en todo el genoma (Tabla 3) intervalos genómicos identificados de este modo se utilizaron para la filtración de los datos de secuenciación del exoma para identificar mutaciones causantes potenciales. Para ser amplia en la identificación de posibles mutaciones causales, tanto en el caso de todo el pedigrí y sub-pedigree los intervalos de la respectiva paramétrica y análisis no paramétricos se combinaron. Para el pedigrí de todo el análisis de esta fue la unión de intervalos descritos en la Tabla 1 junto con los intervalos apropiados en la Tabla 3 (incluyendo 2447) y para el análisis sub-pedigree la unión de intervalos encontrar en la Tabla 2 con intervalos apropiados de la Tabla 3 ( excluyendo 2447). Total exoma secuenciación a continuación realizó la secuenciación del exoma en dos de las tías maternas afectadas para el probando (1826 y 1841) y el primo tercero afectado (2447). Priorizamos y se filtra variantes en función de su frecuencia entre las poblaciones europeas en la ascendencia (< 2% y un filtro más estricto en < 1%) a partir de las 1000 bases de datos de genomas y EXOME proyecto de secuenciación, así como otros exomas secuenciados al mismo proveedor de secuenciación, ubicación dentro de una región genómica de interés, y la consecuencia funcional de la mutación (alteración de la proteína de secuencia o sitio de empalme de transcripción que codifica al menos una proteína de codificación). Las variantes de interés fueron secuenciados por secuenciación de Sanger en otros individuos afectados. filtración variante basada en intervalos genómicas se realizó por separado para cada hipótesis (todo-pedigree reducido el pedigrí) (Tablas S1 y S2). Además de la identificación y la filtración de variantes genéticas, se evaluó la profundidad de la cobertura de la secuenciación de los exones (definido por el conjunto de consenso CDS) dentro de las regiones genómicas de interés ya través de los resultados individuales de secuenciación del exoma. Además, se realizaron búsquedas de variantes que pueden compartirse que fueron "enmascarados" por problemas de cobertura. Para todas las variantes observadas en una o más exomas, si no se observó ninguna variante en el exoma (s) restante, evaluamos si que era debido a vacíos de baja cobertura o la cobertura dentro del exón. Para las variantes, si esto era cierto que filtra utilizando criterios estándar (como el anterior). El uso de estos criterios de filtrado, varias variantes con baja MAF y potencialmente tener un efecto en el nivel de codificación de proteína se observaron en las regiones genómicas de todo el pedigrí de interés que habían sido identificados por análisis de ligamiento paramétrico; Sin embargo, ninguno estaba presente en todos los individuos afectados. Además no hay variantes candidatas "enmascarados" se identifican con los mismos criterios. consideró la posibilidad de que los individuos 1845 y 2447 tienen una condición clínica distinta, y se examinaron las variantes compartidas entre el índice y familia inmediata. Utilizando los mismos criterios de filtrado que el anterior, pero utilizando sólo los individuos exomas de 1826 y 1841, se identificaron un total de 127 variantes. filtrado más estricta para las variantes raras (MAF < 1%) reduce esto a número a 85 (Tabla S2). Un subconjunto de estas variantes, en base a una combinación de factores (mutaciones en COSMIC [13], el efecto de la mutación predijo, la conservación de los aminoácidos codificados, revisión de la literatura, conocidos patrones de expresión en tejidos normales y tumores, fenotipos de la enfermedad asociada con el gen) fueron secuenciados para el seguimiento en el caso índice y de su madre. Una variante en la MAP3K6 gratis (Chr1, NM_004672) fue de particular interés. Una mutación identificada en MAP3K6 gratis (. C [2837C > T]; [=], p.P946L) fue considerado un fuerte candidato en función de las asociaciones conocidas de otros MAP quinasas con cáncer y varias publicaciones para dilucidar una papel para MAP3K6 en la tumorigénesis [10], [11], [14], [15]. Esta variante se ha informado anteriormente (rs141787524) con una frecuencia menor alelo del 0,7% en el Proyecto 1000 Genomas (grupo de descendencia europea) y una frecuencia de 0,4% en la población europea-americana (Exoma Variante Server (NHLBI GO Proyecto Exoma Secuenciación ( ESP):. http://evs.gs.washington.edu/EVS [Consulta de octubre de 2013]) fue visto como una variante heterocigota en 11 (de 1532) otros exomas secuenciados en el Centro de Innovación del Genoma Quebec, que corresponde a una MAF de 0,36% Esta SNV estaba presente en cuatro individuos afectados en la familia Marítima (1884, 1826, 1844, 1841), de los cuales tres mostraron claramente la presencia de células en anillo de sello. Sólo una pequeña biopsia por punción estaba disponible para la tía materna de 1841, por lo tanto, no hemos podido confirmar definitivamente la presencia o ausencia de células en anillo de sello. el MAP3K6 A continuación utiliza el ADN aislado de una sección tumoral de la muestra FFPE del afectado MAP3K6 Verificación de FGC no relacionados Casos Hemos examinado muestras de ADN de un adicional de 115 individuos no relacionados FGC utilizando un multiplexado dirigido siguiente ensayo de secuenciación generación. Las muestras procedían de familias no relacionadas que cumplieron con los criterios internacionales gástrica consorcio vinculación cáncer (IGCLC) para el cáncer gástrico difuso hereditario (106), pero habían probado previamente negativo para la mutación del CDH1 El SNV truncando se observó en un individuo portugués con una historia familiar de cáncer gástrico (Figura 3A). Si bien esto SNV tiene un identificador dbSNP (rs34008139), no hay frecuencia de la población se ha asociado con él de cualquiera de los 1000 genomas o proyectos de servidor Exoma Variant. Este SNV También se ha informado en la base de datos COSMIC (estado somático /línea germinal no especificado) [13] en una muestra de carcinoma del intestino grueso. El examen histológico del tumor del caso índice mostró un cáncer gástrico pobremente diferenciado con células en anillo de sello, conservando expresión de la proteína E-cadherina en la membrana celular (Figura 3B y 3C). La variante p.D200Y se encontró en los probandos de dos familias no relacionadas, y se ha observado dentro de los genomas de cohorte 1000 (rs41291098), sino que también es poco frecuente, con una frecuencia menor alelo (MAF) de 0,4% en ambos grupos de descendencia el 1000 genomas y secuenciación Exoma proyecto europeo. La variante p.P958T (rs75893867) no ha reportado MAF en cualquiera de los 1000 Genomas o exoma secuenciación europeos Proyecto de descenso conjuntos de datos, y sólo fue identificado en los 1000 Genomas en la cohorte japonesa a una frecuencia de 2.2%. Sin embargo, se ha identificado en COSMIC (COSM99077) como una mutación somática a partir de un paciente de carcinoma gástrico. La variante p.V207G ha sido identificado en el Proyecto de Secuenciación del Exoma conjunto de datos europea-americana con un MAF de 0,01%. No se identificó entre los grupos de descendencia europea o europeas dentro de los 1000 Genomas, pero no se observó en otros grupos de población en un rango de frecuencias de alelos menores. Junto con MAP3K6 FFPE tejido tumoral fue disponible para el probando de la familia portuguesa que lleva los p.F849Sfs * 142 de la línea germinal truncando mutación. Las mutaciones somáticas se excluyeron en la secuencia codificante y el exón-intrón completas límites de MAP3K6 Hemos aplicado una variedad de in silico Discusión a continuación se presenta la primera evidencia de que las mutaciones en línea germinal MAP3K6 Proyección de familias adicionales FGC reveló cinco otros MAP3K6 a pesar de MAP3K6
, representan el 40% de la forma más común de cáncer gástrico familiar (FGC), cáncer gástrico difuso hereditario (CGDH). Los genes responsables de las restantes formas de MGF son actualmente desconocidos. Aquí examinamos una familia grande de Canadá marítimo con FGC y sin CDH1
mutaciones, y se identificaron una variante de la línea germinal (p.P946L) en MAP quinasa quinasa quinasa 6 de codificación ( MAP3K6
) . Sobre la base de la conservación, predicho patogenicidad y un papel conocida del gen en la predisposición al cáncer, MAP3K6
se consideró un candidato fuerte y se investigó adicionalmente. Proyección de un 115 individuos no relacionados con adicionales no CDH1
FGC identificó la p.P946L MAP3K6
variante, así como cuatro variantes de codificación adicionales en MAP3K6 gratis (p. F849Sfs * 142, p.P958T, p.D200Y y p.V207G). A-segundo golpear variante somática (p.H506Y) estaba presente en el ADN obtenido a partir de una de las muestras de tumor, y la evidencia de hipermetilación del ADN dentro de la MAP3K6
gen se observó en el ADN del tumor de otro individuo afectado. Estos hallazgos, junto con la evidencia previa de modelos de ratón que MAP3K6
actúa como un supresor de tumores, y los estudios que demuestran la presencia de mutaciones somáticas en MAP3K6
en los cánceres gástricos no hereditarios y celular de cáncer gástrico líneas, punto hacia el MAP3K6
variantes como un factor predisponente para FGC.
. La conservación, predijo la patogenicidad de la variante, la distribución tisular, y la función conocida de MAP3K6
hizo que fuera un candidato fuerte que debe ampliarse la investigación. El examen de un adicional de 115 probandos sin relación identificado mutaciones adicionales en MAP3K6
, incluyendo una mutación truncar
se asocian con el cáncer gástrico familiar. PLoS Genet 10 (10): e1004669. doi: 10.1371 /journal.pgen.1004669
infección crónica tienden a tener mayores cargas de cáncer gástrico [4]. La mayoría de los cánceres gástricos (90%) son esporádicos, pero aproximadamente el 10% muestran la agrupación familiar [5]. Sólo el 1% a 3% son causados por un síndrome hereditario, en oposición a factores ambientales tales como las prácticas dietéticas compartidos [5]. La forma familiar más bien establecida de cáncer gástrico es el cáncer gástrico difuso hereditario (CGDH [MIM 137215]), donde aproximadamente el 40% de los casos se atribuyen a mutaciones en la línea germinal en el gen que codifica E-cadherina, CDH1
[6 ] - [9].
gen. Mientras que la familia tiene muchas características típicas de CGDH, hubo diversidad en la presentación clínica dentro de la familia, así como una avanzada edad de inicio, por lo tanto, se ha optado por referirse simplemente a la condición de FGC sobre el CGDH más estrictamente definido. mapeo genómico de regiones heredadas compartidos entre los miembros de la familia afectados, seguida de la secuenciación del exoma, condujo a la identificación de una variante de la línea germinal de un solo nucleótido (SNV) en MAP quinasa quinasa quinasa 6 ( MAP3K6
, ASK2
, MAPKKK6
, MEKK6
, ENSG00000142733), un gen que codifica un miembro de la familia de la proteína quinasa serina /treonina. Varios in silico
métodos predijeron el SNV en MAP3K6 ser perjudicial para la proteína, y estudios previos con MAP3K6
ratones deficientes [10], así como la aparición de mutaciones en este gen en ambos tumores de cáncer gástrico primarios y líneas celulares de cáncer gástrico [11], fueron consistentes con mutaciones en el MAP3K6
gen que se encuentra la mutación causante. La secuenciación de ADN aislado directamente de una muestra de tumor fijo de un individuo demostrado la presencia de un de novo
segundo-hit variante en MAP3K6
. Proyección de un adicional de 115 muestras de FGC no relacionados, también negativo para CDH1
mutaciones, reveló cinco individuos con cuatro SNVS adicionales en MAP3K6 Windows que también se prevé que ser patógena, así como una persona no relacionada con el SNV identificado en la familia de Canadá marítimo. La edad de inicio varía entre los MAP3K6
portadoras SNV en las cinco familias, y no se había desarrollado cáncer incluso en la fase crónica de la vida, lo que sugiere penetrancia incompleta. Este es el primer informe de un cáncer hereditario que resulta de SNVS en MAP3K6
.
Resultados
fue visto.
.
fue identificado en el fondo de la mucosa gástrica. Una sección pequeña biopsia estaba disponible para un nuevo examen. A pesar de que esta muestra era demasiado pequeña para una clasificación completa, se mostró nidos cohesivos de células neoplásicas con pequeños focos de formación glandular de acuerdo con un adenocarcinoma pobremente diferenciado. Había algunas células tumorales en la muestra que muestran citoplasma claro, pero éstos no se pueden clasificar definitivamente como células en anillo de sello.
y metaplasia intestinal irregular.
no se observó en la mucosa normal adyacente. El tumor fue positivo para dos variantes intrónicas en CDH1
ambos de los cuales se espera que sean benignos (NM_004360.3: c.688-83G > A y c.2439 + 52G > A)
<. p> paciente 2447, un primo tercero, fue diagnosticado a los 44 años con un adenocarcinoma pobremente diferenciado y sin características de células en anillo de sello. La mucosa gástrica adyacente mostró extensa metaplasia intestinal. No hubo evidencia de H. pylori
.
cáncer gástrico no familiar, se añadió una familia sin relación de Portugal con nuestro estudio (Figura 3A). Individuo II-6 fue diagnosticado con cáncer gástrico a los 62 años, que tiene un adenocarcinoma pobremente diferenciado del estómago y la presencia de células en anillo de sello. análisis de inmunohistoquímica mostró positividad de membrana de E-cadherina en las células neoplásicas (Figura 3C), incluyendo las células en anillo de sello (Figura 3B). Los individuos relacionados con I-4, II-1, II-7, y fueron diagnosticados con cáncer gástrico (detalles desconocidos de histología) en edades de 53, 62 y 52, respectivamente. Los cuatro individuos en el pedigrí portugués murieron de la enfermedad dentro de los 5 años del diagnóstico en esta familia. En la familia canadiense Marítimo, 1884, 1844 y 1841 murieron a causa de la enfermedad dentro de un año del diagnóstico.
Mapeo molecular y la Exclusión de genes conocidos y candidatos
, no hay mutaciones en los exones codificantes de proteínas de CDH1
fueron encontrados en los individuos afectados de la familia canadiense Marítima. Para identificar los loci patógena en esta familia, de alta densidad de SNP genotipado utilizando matrices Illumina se realizó en cinco individuos afectados: la madre del probando (1844), dos tías maternas afectados (1826 y 1841), y dos primos lejanos (1845 y 2447) así como varios individuos relacionados, sin incidencia de cáncer cuya condición de afecto fue tratado como desconocido (1907, 1924, 1821, y 1822). Para todas las personas, excepto datos de genotipos 1845 estaba disponible en 2,5 millones de marcadores. Individual 1845 habían sido previamente genotipo a una densidad de 660K, y no era capaz de ser re-genotipo en la densidad más alta. No se obtuvo ADN adecuado para el genotipado SNP de la muestra FFPE del probando (1884). Usando estos datos, hemos realizado tanto no paramétrico y el análisis de ligamiento paramétrico utilizando Merlin [12]. Dada la edad de aparición tardía en muchos miembros de la familia afectados, la penetrancia en el pedigrí marítimo es desconocido. Con el fin de ser conservador en la identificación de regiones genómicas de interés, dos modelos dominantes penetrancia (50% y 99% de penetrancia) utilizando los individuos afectados por 1826, 1841, 1844, y 2447 (e individuos 1907, 1924, 1821 y 1822 con la condición de afecto desconocida ) fueron usados. regiones genómicas identificadas en el análisis de ligamiento paramétrico fueron generalmente consistentes entre sí, independientemente del parámetro elegido penetrancia (Tabla 1). El análisis se repitió con 2447 individuo tratado como desconocido para analizar sólo la reducida pedigrí donde 2447 y 1845 fueron tratados como phenocopies potenciales (Tabla 2). Esto dio lugar a menores LD resultados globales para todas las regiones identificadas, así como más y más grandes regiones, en promedio, que abarca una porción más grande del genoma.
.
.
SNV también estaba presente en cinco de los 27 parientes no afectados actualmente incluidos en la muestra, y se no estuvo presente en el casó en relativo. Uno de los portadores fue homocigotos para el SNV y fue más de 80 años de edad sin cáncer dijo. Aunque no se informó de la consanguinidad en la familia, y no se observó evidencia de la variación del número de copias de los datos de genotipo SNP, este individuo era también homocigotos para una región que abarca 10 Mb locus. Los portadores restantes tenían edades comprendidas entre 33 a 51, y que la edad de aparición del cáncer fue en general más tarde, su estado se consideró "desconocido". Tanto los individuos, 1845 y 2447, con el cáncer gástrico fenotípicamente distinta fueron negativos para el MAP3K6
SNV.
somática variantes dentro del tumor
SNV portador 1884 (el probando) para detectar el SNVS somáticas adicionales o pérdida de heterocigosidad (LOH) en el tumor en sí. Además de la variante p.P946L, hemos identificado un nuevo SNV en el MAP3K6
gen en la posición c [1516C > T]. P.H506Y que conduce a un cambio de aminoácidos (Tabla 4), y fueron capaces de inferir que el SNV fue adquirida somáticamente basado en datos de la secuencia de la cónyuge e hijos.
locus, o cáncer gástrico intestinal familiar. Dentro de este grupo, se identificaron cinco SNVS heterocigóticos adicionales en el MAP3K6
gen (Tabla 4):. Truncando una SNV (. C [2544delC], p.F849Sfs * 142), tres SNVS missense (c [2872C > A], p.P958T;. c [598g > T], p.D200Y; y c [620T >. G], p.V207G), y otro individuo con el p.P946L (c [2837C >. T] ) variante, previamente encontrado en la familia Maritime (Figura 4). Las mutaciones en MAP3K6
fueron descubiertos en los individuos que cumplen los criterios para el cáncer gástrico difuso (106). El individuo en esta novela cohorte de pacientes portadores de la variante p.P946L se cree que es relacionado con la familia Marítimo.
, 50 genes adicionales previamente sugerido que participan en el riesgo de enfermedad del tracto gastrointestinal superior se secuenciaron para esta cohorte usando un ensayo basado en panel personalizado (manuscrito presentado). Los genes fueron seleccionados para la pantalla basada en MiSeq personalizado basado en revisión de la literatura, así como los genes de interés en proyectos de colaboración. En los casos en los que MAP3K6
se identificaron variantes de cambio de sentido, no se encontraron otras variantes candidatos.
-segundo golpe Análisis en el tumor de la familia portuguesa
en el tumor del individuo afectado. La falta de LOH en el MAP3K6
gen también podría inferirse en este tumor, ya que tanto de tipo salvaje y alelos mutantes pudieron ser identificados en el sitio de la mutación de la línea germinal (Figura S2). Por lo tanto, se realizaron búsquedas de mecanismos alternativos de inactivación putativos. La hipermetilación de las islas CpG dentro de los promotores de genes y regiones reguladoras es un fenómeno común que conduce a la disminución de la expresión de genes en el cáncer [16]. MAP3K6
regulación por hipermetilación del promotor se ha descrito para el hueso humano de células madre mesenquimales de la médula [17], aunque no se ha establecido una correlación de la hipermetilación y la expresión génica. Se realizaron búsquedas de MAP3K6
islas CpG [18] y encontró dos islas CpG, uno en la región promotora y otro que abarca el exón 10 y parte del intrón aguas abajo (Figura 5 y Figura complementario S1). La isla CpG aguas abajo (isla CpG 2) está cerca de un sitio hipersensible DNasa predice para albergar promotor asociado características (función reguladora Ensembl ID ENSR00000533270, Figura 5). Nos ADN tratado con bisulfito de: linfocitos de sangre periférica del individuo afectado (PBL); El tumor de la persona afectada; cuatro diferentes muestras de control normal del estómago, y; siete líneas celulares de cáncer gástrico. Para la isla CpG promotor (isla CpG 1), no se detectó hipermetilación utilizando dos conjuntos de cebadores diferentes (Figura 5 y Supl. Figura S1). En cuanto a la isla CpG 2, se observó metilación completa para el ADN del tumor y no metilación para el ADN PBLs. Curiosamente, el análisis de metilación en la isla de CpG 2 en la mucosa normal del estómago de los controles mostró un patrón de metilación parcial. En línea con el resultado obtenido para el ADN del tumor, las siete líneas celulares de cáncer gástrico muestran metilación completa (Figura 5 y Supl. Figura S1).
Patogenicidad de SNVS en MAP3K6
métodos para predecir la patogenicidad de los SNVS missense observados. Aunque no hubo consenso total entre los programas (tabla 5), todos los SNVS fueron considerados perjudiciales por lo menos un programa, y salvo p.V207G y p.P958T, las otras cuatro variantes descritas en este informe se prevé que ser nocivo por al menos 3 de los 7 métodos. Además, el EvoD [19] consenso de predicción (basado en una combinación equilibrada de la EvoD, PolyPhen2 [20], y SIFT [21] puntajes) informó que tres de las variantes (p.D200Y, p.V207G, p.H506Y ) eran perjudiciales o que puedan ser perjudiciales. Las dos variantes restantes, p.P946L y p.P958T, se prevé que sea probable que los cambios neutros aunque eran evolutivamente ultra-conservados y bien conservados (según la clasificación de la tasa de evolución EvoD), respectivamente; mientras que sólo una de las otras tres variantes (p.D200Y) fue considerado bien conservado y los otros dos fueron menos conservadas. Como (i) un fue encontrado MAP3K6
truncar variante, (ii) se produjo un segundo golpe variante identificada en el individuo para el cual se examinó el tejido FFPE, y (iii) todos los programas de prueba están diseñados para patogenicidad predecir sobre la base de la pérdida de la función, es probable que las variantes descritas daría lugar a un descenso de la función o el fenotipo dominante negativo.
están relacionados con el cáncer hereditario. Se encontraron cuatro individuos con cáncer gástrico de una familia canadiense Marítima para realizar una variante heterocigota en el MAP3K6
gen, lo que lleva a un cambio de aminoácido p.P946L. Esta variante de la línea germinal, localizado en el cromosoma 1, se identificó en dos de las tres muestras exoma y se encuentra dentro de una región identificada por el análisis paramétrico y no paramétrico vinculación dentro de la sub-pedigree (1845 y 2447 tratados como estado de la enfermedad "desconocido") . La importancia de la MAP3K6
variante fue apoyada por la identificación de un segundo golpe mutación somática en el MAP3K6
gen en p.H506Y presentes en el ADN aislado directamente de una sección tumoral de un paciente FFPE muestra. Dos individuos del pedigrí con cáncer gástrico, pero con algunas diferencias fenotípicas no portaban la mutación; Sin embargo, no hay variantes candidatos fueron identificados comparten entre todos los individuos afectados dentro de cualquier región identificada, por ligamiento paramétrico llevaron a cabo en todo el pedigrí.
mutaciones, incluyendo una p.F849Sfs * 142 de la línea germinal mutación observada en el probando portugués, que se espera que conduzca al truncamiento de proteínas. Después de excluir las mutaciones somáticas y LOH, un posible mecanismo de segundo golpe se encontró a través de
hipermetilación en una isla CpG intragenic cerca de un elemento regulador promotor asociado predicho (DNasa I hipersensible sitio). La relevancia de la metilación en este MAP3K6
región del gen podría no determinarse en términos de impacto en la expresión de genes, sin embargo, la posibilidad de actuar como una posible inactivación segundo mecanismo de golpear (parcial o completa) se eleva, debido a las los resultados obtenidos en líneas celulares de cáncer de estómago y normales. Si es así, esto puede representar otro ejemplo del concepto reconoce cada vez más que la metilación del ADN en el cuerpo de genes no es sólo un testigo pasivo de la transcripción de genes, sino que participa activamente en varios procesos de regulación de genes [22], lo que justifica una mayor investigación. El análisis histopatológico de la persona de la familia portuguesa que lleva esta mutación truncar contó con células en anillo de sello como parte del fenotipo tumoral, al igual que la mayoría de los individuos de la Marítima sub-pedigree (a excepción de 1841, en el que el estado del anillo de sello no fue concluyente debido a la falta de material suficiente).
mutaciones no han sido previamente identificados en el cáncer hereditario, hay una creciente evidencia de que MAP3K6
tiene un papel importante en la patogénesis del cáncer . En los ratones, donde MAP3K6
normalmente se expresa en el tejido gástrico y de la piel, la pérdida de MAP3K6 Hoteles en ratones knockout homocigotos se encontró para aumentar la susceptibilidad al cáncer de piel inducido por [10]. Los ratones no desarrollan cáncer de forma espontánea; sin embargo, la inducción química realizado en presencia de un estímulo inflamatorio condujo a un mayor número de tumores de la piel en el MAP3K6
ratones deficientes que en los animales de control. El número de tumores en los heterocigóticos ( MAP3K6
+/-) ratones, así como su tamaño, fue intermedia entre la de tipo salvaje y ratones knock-out, lo que sugiere un papel para MAP3K6
dosificación en sus efectos [10].
 Una variante genética común explica por qué la inmunoterapia a menudo falla en la enfermedad de Crohn
Una variante genética común explica por qué la inmunoterapia a menudo falla en la enfermedad de Crohn
 Aumento de los riesgos de biodefensa que plantea la biología sintética
Aumento de los riesgos de biodefensa que plantea la biología sintética
 Bacterias intestinales relacionadas con cambios metabólicos y autismo en un nuevo estudio
Bacterias intestinales relacionadas con cambios metabólicos y autismo en un nuevo estudio
 El microbioma vaginal puede afectar la eficacia de la terapia de prevención del VIH
El microbioma vaginal puede afectar la eficacia de la terapia de prevención del VIH
 La proteína SARS-CoV N provoca la producción de IFN-β al provocar la ubiquitinación de RIG-I,
La proteína SARS-CoV N provoca la producción de IFN-β al provocar la ubiquitinación de RIG-I,
 Diabetes tipo 1 relacionada con el microbioma intestinal y factores genéticos
Diabetes tipo 1 relacionada con el microbioma intestinal y factores genéticos
 El extracto de semilla de xilitol y pomelo se muestra prometedor para prevenir la infección por SARS-CoV-2,
hallazgos del estudio La enfermedad del coronavirus (COVID-19), causado por el síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2), ha causado estragos en todo el mundo. Se ha extendido desd
El extracto de semilla de xilitol y pomelo se muestra prometedor para prevenir la infección por SARS-CoV-2,
hallazgos del estudio La enfermedad del coronavirus (COVID-19), causado por el síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2), ha causado estragos en todo el mundo. Se ha extendido desd
 La terapia biológica puede disminuir el riesgo de COVID-19 grave
Un estudio observacional reciente realizado por investigadores españoles, actualmente disponible en el medRxiv * servidor de preimpresión, sugiere que los pacientes con enfermedades inmunomediadas q
La terapia biológica puede disminuir el riesgo de COVID-19 grave
Un estudio observacional reciente realizado por investigadores españoles, actualmente disponible en el medRxiv * servidor de preimpresión, sugiere que los pacientes con enfermedades inmunomediadas q
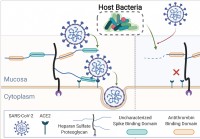 El microbioma humano recorta los glucanos de las mucosas,
que influye en la infección por SARS-CoV-2 Un equipo internacional de investigadores ha realizado un estudio que muestra que las diferencias en el microbioma humano pueden influir en la capacidad del
El microbioma humano recorta los glucanos de las mucosas,
que influye en la infección por SARS-CoV-2 Un equipo internacional de investigadores ha realizado un estudio que muestra que las diferencias en el microbioma humano pueden influir en la capacidad del