Extracto
Antecedentes
La evidencia acumulada indica que la metilación aberrante del ADN está implicado en la génesis de tumores gástricos, lo que sugiere que puede ser una biomarcador clínica útil para la enfermedad. El objetivo de este estudio fue consolidar y resumir los datos publicados sobre el potencial de metilación en el cáncer gástrico (CG) de predicción de riesgo, pronóstico y predicción de respuesta al tratamiento.
No se identificaron estudios pertinentes PubMed utilizando un enfoque de búsqueda sistemática. Los resultados se resumieron mediante el metanálisis. Mantel-Haenszel odds ratio se calcularon para cada evento metilación asumiendo el modelo de efectos aleatorios.
Una revisión de 589 publicaciones recuperados identificó 415 artículos pertinentes, incluyendo 143 estudios de casos y controles sobre el gen metilación de 142 genes individuales en muestras clínicas de GC. Un total de 77 genes fueron significativamente metilado diferencialmente entre tumor y el tejido gástrico normal a partir de materias de GC, de los cuales los datos sobre 62 se derivan de los estudios individuales. La metilación de 15, 4 y 7 genes en Normal gástrico tejido, plasma y suero, respectivamente, fue significativamente diferente de la frecuencia entre los sujetos de GC y no cancerosas. Un significado pronóstico se informó de 18 genes y la significación predictiva se informó de p16 metilación del ADN es un biomarcador prometedor para GC la predicción del riesgo y el pronóstico. Se requiere la validación más enfocada de marcadores de metilación candidatos en cohortes independientes para desarrollar su potencial clínico Visto:. Sapari NS, Loh M, Vaithilingam A, Soong R (2012) potencial clínico de la metilación del ADN en el cáncer gástrico: Un El meta-análisis. PLoS ONE 7 (4): e36275. doi: 10.1371 /journal.pone.0036275 Editor: Rossella Rota, Ospedale Pediatrico Bambino Gesú, Italia | Recibido: Septiembre 29, 2011; Aceptado: March 31, 2012; Publicado: 27 Abril 2012 Derechos de Autor © 2012 Sapari et al. Este es un artículo de acceso abierto distribuido bajo los términos de la licencia Creative Commons Attribution License, que permite el uso ilimitado, distribución y reproducción en cualquier medio, siempre que el autor original y la fuente se acreditan Financiación:. Este trabajo el apoyo de una beca del Consejo Nacional de Investigación médica (NMRC /TCR /001/2007). Los donantes no tenía papel en el diseño del estudio, la recogida y análisis de datos, decisión a publicar, o la preparación del manuscrito Conflicto de intereses:.. Los autores han declarado que no existen intereses en competencia Introducción el cáncer gástrico (CG) sigue siendo un problema clínico importante en todo el mundo debido a su alta prevalencia, mal pronóstico y las opciones de tratamiento limitadas [1]. Aunque la incidencia de GC ha disminuido a lo largo de los años, sigue siendo la segunda causa principal de muerte por cáncer y el cuarto cáncer más común en todo el mundo. Menos del 25% de los casos de CG son diagnosticados en una etapa temprana, y la tasa de supervivencia a 5 años es sólo del 24% en los EE.UU. y Europa [2]. Sin embargo, la tasa de supervivencia de GC mejora de más del 60% si se detecta en una etapa temprana [2], haciendo hincapié en la importancia de la detección precoz de este tipo de cáncer. metilación del ADN es un mecanismo epigenético de la regulación transcripcional, con una participación en el cáncer atribuido al silenciamiento inadecuado de los genes supresores de tumor, o la pérdida de la represión oncogén [3]. Desde el primer artículo de Fang et al. El patrón de metilación del ADN del tumor puede ser útil para la detección del riesgo de cáncer, el pronóstico y el tratamiento de predicción [3], [ ,,,0],26] - [30]. En comparación con la mutación somática, la metilación del ADN tiene un mayor número de alteraciones aberrantes por célula del cáncer [31]. Por otra parte, la metilación aberrante del ADN se produce a principios de la génesis de tumores de muchos tipos de cáncer [28], por lo que es particularmente útil para la predicción del riesgo. La atracción técnica de la metilación del ADN es que es químicamente estable y puede detectarse con una sensibilidad muy alta de hasta 1:1000 moléculas [19]. Varios informes han demostrado también que, ADN metilado cáncer específica se puede encontrar en los fluidos biológicos, lo que sugiere que podría ser un marcador útil para el diagnóstico no invasivo [28], [32], [33]. La importancia de la detección precoz para mejorar la supervivencia de los resultados de GC y la evidencia prometedora de la metilación del ADN como biomarcadores es la motivación para este estudio. A pesar de la creciente evidencia del potencial clínico de la metilación del ADN, muchas inconsistencias en los resultados se pueden observar en todos los estudios. Por lo tanto, este estudio se realizó para consolidar la información sobre el potencial clínico de metilación en GC por medio de un meta-análisis, y sugerir cuál de los candidatos eventos de metilación merecen una evaluación posterior como biomarcadores clínicamente relevantes para la enfermedad. identificación y elegibilidad de los estudios Una búsqueda sistemática de la literatura en PubMed de artículos publicados hasta al 27 de octubre de 2011 es realizado usando ' "cáncer gástrico" y "metilación"' como la búsqueda condiciones. No se utilizaron restricciones durante la búsqueda en PubMed y los estudios resultantes fueron manualmente curada de acuerdo a su relevancia para la metilación del ADN GC. Estos estudios incluidos de GC en las áreas de la hipermetilación y la hipometilación /desmetilación de las regiones globales y específicos de la diana. El título y el resumen de los artículos identificados en la búsqueda inicial fueron evaluados para comprobar su adecuación a los objetivos de este documento. Todos los artículos potencialmente relevantes fueron evaluados en detalle y se identificaron estudios adicionales pertinentes, de las citas dentro de estos artículos. Dado que el objetivo de este trabajo fue en la metilación de genes humanos, los estudios que analizaron la metilación de H. Opiniones y pylori Epstein-Barr Selección de los estudios y la anotación de datos El meta-análisis de frecuencias se resumen en el tumor y el tejido gástrica normal se limita a los datos de los estudios de casos y controles que informaron la frecuencia de metilación de genes individuales en los grupos respectivos. Los datos de revisiones y metanálisis de los mismos estudios no fueron consideradas. La información sobre el primer autor, año de publicación, el gen (s) analizada, el tamaño de la población de estudio, la frecuencia de casos y controles metilados, los métodos utilizados para el análisis de metilación del ADN y el tipo de muestra se registran para cada estudio (Tabla S1, S2). Otros detalles relevantes tales como el tipo de lesión, H. pylori Los meta-análisis se realizaron utilizando el Review Manager 5 (The Cochrane Collaboration, Copenhague, Dinamarca). Mantel-Haenszel odds ratios (OR) se calcularon para cada gen mediante la aplicación del modelo de efectos aleatorios. La homogeneidad entre los estudios para el mismo gen se evaluó sobre la base de la χ 2 test mediante la estadística Q de Cochran. La estadística de I 2, que mide el grado de inconsistencia entre los estudios, se evaluó también. Se utilizó el intervalo de confianza del 95% de la odds ratio para evaluar las diferencias entre los grupos. Si los intervalos de confianza no se solapan, los dos odds ratios fueron significativamente diferentes al nivel del 10% (desde el 1- (0,95 * 0,95) ≈0.9). gráficos de embudo, así como pruebas de Begg se utilizaron para comprobar si hay sesgo de publicación [34]. El sesgo de publicación se consideró significativa cuando el valor de p fue. < 0,1 [35] Resultados Características del estudio El proceso de selección de artículos utilizados en este estudio se resume en Figura 1. Un total de 559 estudios se identificaron en PubMed, y otros 30 estudios adicionales se identificaron más lejos de las citas de las publicaciones iniciales recuperados. Sobre la base de la idoneidad del título y el resumen de los objetivos del estudio, se seleccionaron 415 artículos para su posterior evaluación detallada. De éstos, se excluyeron 190 estudios de casos y controles no, 22, 4 críticas 1 comentarios y meta-análisis de casos y controles metanálisis. También se excluyeron otros estudios de casos y controles, como lo fueron los estudios de que no son proteínas los genes de codificación (por ejemplo, la metilación de los genes de micro-ARN), la presentación de los datos no era adecuado (por ejemplo, datos de los sitios CpG, y desmetilación o hipometilación solamente) o los datos de frecuencia era deficiente. En total, se consideraron 143 estudios de casos y controles que informaron la frecuencia de metilación de 142 genes individuales para los metanálisis. se utilizaron los datos de algunos estudios en más de un meta-análisis, ya que contenían datos sobre múltiples comparaciones de tipos de muestras consideradas en este estudio. Los genes metilados diferencialmente entre el tumor y el tejido gástrica normal de sujetos con cáncer gástrico se identificaron un total de 106 estudios de casos y controles que informaron sobre la frecuencia de metilación en 122 genes en muestras de tejido normal y tumoral de sujetos GC para el metanálisis (Tabla S1). La metilación de 77 de los 122 genes fue significativamente diferente entre las muestras (Tabla 1), de los cuales los datos de 62 se deriva de un único estudio. La metilación fue significativamente mayor en los tumores en 70 genes, y en el tejido normal en 7 genes. Veinte estudios de casos y controles comparando la frecuencia de metilación de genes entre 34 muestras de tejido normal de los sujetos GC y no cancerosas se identificaron para el metanálisis (Tabla S2). De éstos, la metilación en 15 genes fue significativamente diferente, de los cuales los datos de 11 genes se obtuvieron a partir de un único estudio sólo (Tabla 2). Para todos los 15 genes, la metilación fue mayor en tejido normal de GC en comparación con sujetos no cancerosas. Teniendo en cuenta los eventos de metilación examinados en más de un estudio, 4 ( p16 Los genes diferencialmente metilados en muestras no tejidos de sujetos GC y no cancerosas Un total de 26 estudios que informan sobre la metilación en 29 genes en muestras clínicas diferentes de tejido gástrico, incluyendo sangre entera , plasma, suero, lavados gástricos, se identificaron las muestras de fluidos y fecales peritoneales de los sujetos GC. De éstos, 13 estudios que examinaron la metilación de un total de 14 genes, ya sea en suero, plasma o muestras fecales eran de un diseño de casos y controles y por lo tanto adecuado para el metanálisis (Tabla S2). Significativamente se observaron diferentes frecuencias de metilación en 4 genes en muestras de plasma, 7 genes en muestras de suero y 0 en las heces (Tabla 2). p15 la metilación como marcador pronóstico y predictivo de GC se identificaron un total de 28 estudios que investigaron la metilación de genes en 40 genes en relación con el resultado de supervivencia de los sujetos GC (Tabla 3). De los 40 genes estudiados, sólo el 5 se examinaron en múltiples estudios. Los meta-análisis no pudo realizarse en esta serie de estudios debido a la notificación irregular de los coeficientes de riesgo. Existe una asociación significativa con la supervivencia se informó de metilación en 18 de los 40 genes (45%), aunque se observó inconsistencia en el hallazgo de diferencias significativas para todos los genes examinados en múltiples estudios Cinco estudios informaron sobre las asociaciones entre la supervivencia en sujetos GC recibe un tratamiento de quimioterapia específica y la metilación de genes, incluyendo 10 TMS1 Efecto de la variabilidad y el sesgo de publicación analítica Durante la anotación de los estudios, se observó una heterogeneidad considerable en muchos parámetros del estudio, incluyendo los ensayos utilizados, sitios CpG interrogado, etapas de la enfermedad examinada, y el tipos de muestras utilizadas (por ejemplo, fresco, congelado o tejido incluido en parafina). Doce ensayos diferentes se utilizaron para la evaluación de la metilación, con PCR específica de metilación (MSP) es la más común. Para examinar la influencia de ensayo de metilación en los resultados del estudio, los datos de metilación específica de PCR y el análisis cuantitativo-PCR específica de metilación (por ejemplo, MethyLight) se compararon para el gen examinado con mayor frecuencia en los estudios que comparan la metilación entre tumor gástrico y el tejido normal ( MLH1 Para probar el sesgo de publicación, los datos a partir de los mismos 13 estudios sobre también se examinó MLH1 numerosos estudios han implicado a la metilación del ADN aberrante en numerosos genes en diferentes muestras y modelos de génesis tumoral gástrico [1], [5] - [10], [12] - [14], [16], [18] - [22] , [24], [43]. Esta implicación ha su vez dado lugar a la idea de que la metilación podría ser un biomarcador útil para mejorar el manejo clínico de GC [11], [15], [32], [44], [45]. Hasta la fecha, sin embargo, este potencial no se ha realizado, presumiblemente debido a la falta de pruebas pertinentes para apoyar los ensayos de metilación en la clínica. En este estudio, un examen exhaustivo de todas las publicaciones sobre las frecuencias y las asociaciones de metilación en muestras clínicas de cáncer gástrico se realizó para consolidar la información en el campo. Se realizaron metanálisis cuando sea posible para obtener un consenso objetiva de hechos investigados en varias ocasiones. A partir del análisis, las listas se generaron de genes significativamente diferencialmente metiladas entre tumor y muestra de tejido normal a partir de sujetos GC (Tabla 1), y el tejido normal y /o sangre de GC y los sujetos no cancerosas (Tabla 2), cada uno con eventos de metilación anotado por su fuerza de asociación y la frecuencia de los análisis. Los resultados de los estudios sobre el significado pronóstico y predictivo de eventos de metilación también fueron revisados (Tabla 3). Estas listas y datos complementarios adicionales (Tablas S1, S2) deben proporcionar información útil a partir de la cual evaluar mejor el potencial clínico de los respectivos eventos, y dar prioridad a seguir trabajando Consta de 77% (101/132) de los casos -Control análisis (Figura 1), el mayor grupo de estudios revisados fueron aquellos que compararon la frecuencia de metilación en el tumor y el tejido gástrico normal a partir de materias de GC. La revisión identificó 77 eventos significativos de metilación de genes, lo que confirma mediante meta-análisis, al mismo tiempo significativamente diferente de la metilación de un número de genes implicados en la génesis tumoral comúnmente, incluyendo MLH1 a partir de los estudios que comparan los niveles de metilación en el tejido normal, plasma y suero de GC y no cancerosas sujetos, 15, 4 y 7 (Tabla 2) significativamente diferentes eventos de metilación de genes, respectivamente, se identificaron. Los genes con ambos roles establecidos (como p16 la hipótesis de que el silenciamiento génico por metilación también puede determinar la gravedad de la enfermedad [27] también ha impulsado numerosas investigaciones de las asociaciones de metilación del gen con la supervivencia en GC. En esta revisión, también se identificaron 28 estudios que informan sobre la asociación de la supervivencia de los sujetos GC y metilación en 40 genes. En apoyo de esta hipótesis, se registraron numerosas asociaciones significativas entre la metilación y la supervivencia de los pobres, sobre todo en los genes supresores de tumores (Tabla 3). También se informó de las asociaciones entre la metilación y la mejor supervivencia de cuatro genes ( PTGS2 Los datos de cinco estudios en pacientes que reciben quimioterapia también se ha sugerido que la metilación del ADN en CHFR Los resultados de este estudio ponen de manifiesto un potencial prometedor para la metilación del ADN en la predicción del riesgo de CG, pronóstico y predicción de la respuesta al tratamiento. Sin embargo, muchas cuestiones pertinentes a la aplicación clínica no se abordan en los estudios. Metodológicamente, los estudios definen adecuadamente los enfoques más adecuados para el análisis, debido a su gran variabilidad en los ensayos de PCR, cebadores y sondas, las condiciones de PCR, y los umbrales de positividad utilizan. La mayoría de los estudios (102/143, 71%) se han basado en PCR específica de metilación, para la cual la naturaleza no cuantitativa de análisis presenta dificultades para el control de la calidad y estandarización. Con metilación un evento dinámico, los protocolos para el muestreo también están en necesidad de clarificación, tanto con respecto a la región o el sitio de toma de muestras, y el tiempo de muestreo. La distancia del tejido normal del tumor [46], y el tiempo de muestreo [21], [22] han sido todos documentado para influir de manera significativa los niveles de metilación. La gran variabilidad de los genes y los paneles de genes examinados entre los estudios , combinado con la falta de validación en serie independiente y caracterización de las características de rendimiento de ensayo, también hace que sea difícil definir una prueba clínicamente relevante. La variación en el interrogatorio de frecuencia funcionalmente diferentes sitios CpG [47] - [49] entre los estudios del mismo gen ofrece complicaciones adicionales. Por otra parte, todos los eventos de metilación de genes examinados en múltiples estudios y significativo asociado con GC (incluyendo p16 mayor consideración son las covariables de análisis que serían analizadas con la metilación. La metilación se ha asociado con muchas características demográficas, clínicas y moleculares, incluyendo la edad, el sexo, el tabaquismo, la metaplasia intestinal, la genética del huésped, y H. pylori En conclusión, los resultados de este estudio se resumen un valor prometedor para la metilación del ADN a la predicción del riesgo, el pronóstico y la predicción de la respuesta a la quimioterapia de GC. Sin embargo, importantes cuestiones metodológicas y validación siguen pendientes de resolución para proporcionar los datos que permitirá a esta información para ser considerado para la clínica. Esto incluye el análisis de las series con mayor muestra independiente, la aplicación de métodos normalizados, el ajuste de las covariables en el análisis multivariante, una mayor definición de los criterios de valoración de resultados y de ajuste por el efecto de la intervención terapéutica. La realización del potencial de la metilación del ADN a GC manejo clínico espera de su resolución. Reconocimientos Agradecemos a Barry Iacopetta y Chee Seng Ku por su contribución a la elaboración del manuscrito.
metilación, aunque también se observaron muchos resultados inconsistentes. Sin sesgo debido a ensayo, el uso de tejido fijado o sitios CpG analizadas se detectó, sin embargo se observó un ligero sesgo hacia la publicación de los resultados positivos.
Conclusiones
En 1996 que describe la hipometilación del ADN de c-myc y c-Ha-ras en GC [4], más de 550 se han publicado estudios sobre la participación de la metilación aberrante del ADN en el desarrollo de GC. Como resultado, se ha reportado la presencia y consecuencias funcionales de la metilación del ADN aberrante de más de 100 genes en GC [5] - [17]. La evidencia sobre los vínculos entre la metilación aberrante del ADN a H. pylori
infección [1], [18] - [22] y su participación en las lesiones epiteliales gástricas precancerosas y GC progresión [18], [19], [21], [23] - [25] también están siendo cada vez documentado. Tomados en conjunto, estos resultados han indicado la metilación del ADN aberrante tiene un papel significativo en el desarrollo del cáncer gástrico y la progresión.
Materiales Métodos y
virus (EBV) genomas en la progresión del GC no fueron considerados.
estado, clasificación de Lauren y la isla CpG methylator fenotipo de estado (CIMP) también se registraron cuando estén disponibles. Los casos clínicos se agruparon en ocho categorías, a saber, (1) la mucosa normal de sujetos no cancerosas, (2) acorde con mucosa normal de casos con tumor, (3) la gastritis crónica, (4) metaplasia intestinal, (5) adenoma displásico, ( 6) adenocarcinoma, (7) GC temprano y (8) GC avanzada. Debido al pequeño número de estudios disponibles, los casos clínicos no se clasifican además en H. pylori
casos positivos /negativos o sujetos intestinal /difusa tipo GC y no cancerosas no se clasifican en los controles emparejados /inigualables. Por la misma razón, no se realizó un análisis de subgrupos en función de las diferentes etapas de precancerosa a lesiones cancerosas. Por razones de coherencia, un solo gen nombre basado en la nomenclatura HUGO fue asignado a los genes con múltiples denominaciones.
Metanálisis
Los genes diferencialmente metilado en tejido gástrico normal desde GC y no cancerosas sujetos
, CDH1
, DAPK
, CHFR
) resultaron ser significativamente diferente .
era un gen común identificados en los estudios de plasma y suero, lo que es 10 genes únicos en conjunto significativamente diferentes en la frecuencia de metilación en muestras de sangre. La metilación de sólo dos genes ( CDH1, p16
) fue examinado en más de un estudio, y ambos fueron significativamente diferentes en frecuencia en las muestras de GC y los sujetos que no tienen cáncer de meta-análisis.
.
, DAPK
, LOX
, MGMT
y CHFR
[36] - [40]. En los 5 estudios, los sujetos con metilación tuvieron una supervivencia peor que los no metilación. Un estudio examinó las diferencias de supervivencia entre los pacientes tratados con quimioterapia o sin acuerdo con estado de metilación de p16
[41]. En este estudio, los sujetos sin p16
metilación que recibieron quimioterapia tuvieron una mejor supervivencia que aquellos que no lo hicieron, mientras que para los sujetos con p16
metilación, no hubo diferencias significativas en función del estado del tratamiento.
, 13 estudios de casos y controles). No hubo diferencia estadística ( P Hotel < 0,05) en los intervalos de confianza del 95% de las frecuencias de metilación reportaron el uso de los dos ensayos (Figura S1). No se observaron diferencias significativas en función del tipo de muestra (congelados vs tejido incluido en parafina), estadio de la enfermedad, o en los sitios CpG interrogado también fueron observados en estos estudios (resultados no mostrados).
metilación mencionado anteriormente. Se observó una tendencia hacia una imagen positiva en las gráfico en embudo (Figura S2) y el análisis de la prueba de Begg. Sin embargo, estos resultados deben ser interpretados con cautela, ya que la mayoría de los estudios fueron de tamaño pequeño de la muestra, y un mínimo de 20 estudios generalmente se recomienda para un análisis fiable de sesgo de publicación [42].
Discusión
.
, p16
, y CHFR
y RUNX3 gratis (Tabla 1). Estos eventos representan herramientas útiles para una mejor comprensión tumourigenesis gástrico y, potencialmente, la identificación de nuevas estrategias terapéuticas [3], [28]. Desde la perspectiva de los marcadores de riesgo Sin embargo, estos eventos sólo pueden considerarse un primer grupo de candidatos para poner a prueba aún más en los análisis más clínicamente relevantes, como los eventos en su propia sólo identifican muestras de tumores gástricos que ya son histológicamente diagnosticable.
, CDH1
, DAPK
, RUNX3
, p15
) y en menor papeles: conocidas (como BX161496
, SULF1
, RPRM
) en la génesis de tumores fueron identificados. Una serie de eventos fueron en común entre los estudios sobre el tejido normal y la sangre, como la metilación en p16
, CDH1
, DAPK
. Estos eventos son clínicamente prometedor, ya que demuestran capacidades discriminativas para estimar el riesgo de GC a partir de muestras que se pueden obtener en la práctica habitual actual, como por ejemplo durante el cribado endoscópico (tejido), o de la población o el cribado clínico (sangre).
, MINT31
, MLH1
, MAL
), es de suponer que refleja que la supresión de la actividad oncogénica de la metilación de genes también puede ocurrir. Sin embargo, una considerable falta de estudio independiente y la replicación de las asociaciones para la mayoría de los genes estudiados (Tabla 3) pone de relieve la necesidad de una mayor investigación en este aspecto de la metilación en GC.
, DAPK
, TMS1
podría ser predictores útiles de respuesta a la quimioterapia [36] - [40]. Sin embargo, desde el diseño de estos análisis, es difícil determinar si las diferencias de supervivencia fueron debido a las diferencias inherentes de pronóstico o eran una función de una interacción tratamiento, o ambos. En un estudio de un diseño diferente, Mitsuno et al.
Informó que los pacientes con metilación de p16 obtuvieron un beneficio de supervivencia de la quimioterapia, mientras que los que no tienen la metilación no [41] hicieron. Este resultado sugiere que la metilación de p16 puede ser un marcador útil para predecir la respuesta a la quimioterapia, y proporciona evidencia de una interacción tratamiento. Sin embargo, este estudio sólo examinó 56 sujetos en un análisis retrospectivo, y se requiere mucho trabajo adicional para confirmar estos hallazgos.
, DAPK
, CHFR
, MLH1
, RUNX3
) han sido implicados como marcadores de riesgo de muchos otros tipos de cáncer [26], [27], [50], que plantea dudas sobre la interpretación de su detección en individuos asintomáticos
.
y Epstein Barr virus de estado [1], [24]. Además de las asociaciones directas, muchos estudios también han informado de la modificación de las interacciones entre muchas de estas características y metilación en riesgo de CG [51]. eventos de metilación mismos también pueden estar vinculados e interactúan entre sí [50], que presenta un reto para definir un panel óptima de marcadores de metilación también. Una isla CpG methylator fenotipo (CIMP), que consta de distintos subtipos de GC con los patrones de metilación coordinados, se ha descrito [52] - [55], aunque la evidencia de CIMP en GC no es tan convincente como para el cáncer colorrectal [56 ], [57].
Información de Apoyo
figura S1.
Bosque-parcela de estudios comparando metilados MLH1
metilación entre el tumor y el tejido normal de los sujetos GC de acuerdo con el uso de la PCR específica de metilación y la PCR específica de metilación cuantitativa.
doi: 10.1371 /journal.pone.0036275.s001 gratis (TIF)
figura S2.
embudo parcela de los 13 estudios de MLH1
metilación en el tumor y el tejido gástrico normal a partir de materias de GC para evaluar el sesgo de publicación. La línea vertical indica la estimación combinada de la OR global y las líneas inclinadas representan el intervalo de confianza del 95%
doi:. 10.1371 /journal.pone.0036275.s002 gratis (TIF)
Tabla S1. List de los genes metilados y sus estudios componentes para comparar las diferencias entre tumor y el tejido gástrica normal de sujetos con cáncer gástrico. relaciones de meta-análisis de probabilidad (OR) y el intervalo de confianza del 95% se calcularon para todos los genes metilados analizados. negrita de color rojo y se utilizan para indicar diferencias significativas entre los grupos. BS-SSCP: bisulfito de polimorfismo de conformación de cadena sencilla; Bseq: secuenciación de bisulfito; COBRA: combina el análisis de restricción de bisulfito; DPHLC: desnaturalización cromatografía líquida de alta; FFPE: parafina fijado en formol e incrustados; HRM: alta resolución de fusión; MSP: reacción en cadena de la polimerasa específica de metilación; PCR: reacción en cadena de la polimerasa; QMSP:. Reacción en cadena de la polimerasa específica de metilación cuantitativa
doi: 10.1371 /journal.pone.0036275.s003 gratis (XLS)
Tabla S2. List de los genes metilados y sus estudios componentes para comparar diferencias en el tejido normal, plasma y suero entre el cáncer gástrico y sujetos que no tienen cáncer. relaciones de meta-análisis de probabilidad (OR) y el intervalo de confianza del 95% se calcularon para todos los genes metilados analizados. negrita de color rojo y se utilizan para indicar diferencias significativas entre los grupos. COBRA: combina el análisis de restricción de bisulfito; MSP: reacción en cadena de la polimerasa específica de metilación; QMSP:. Reacción en cadena de la polimerasa específica de metilación cuantitativa
doi: 10.1371 /journal.pone.0036275.s004 gratis (XLS)
 Garrapatas que ahora son portadoras de múltiples enfermedades,
Garrapatas que ahora son portadoras de múltiples enfermedades,
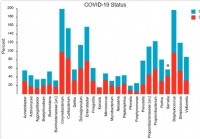 La composición y estructura del microbioma nasofaríngeo se relacionan con la gravedad de la enfermedad COVID-19
La composición y estructura del microbioma nasofaríngeo se relacionan con la gravedad de la enfermedad COVID-19
 Los plásticos ahora se encuentran comúnmente en las heces humanas
Los plásticos ahora se encuentran comúnmente en las heces humanas
 La enfermedad del intestino irritable aumenta el riesgo de demencia
La enfermedad del intestino irritable aumenta el riesgo de demencia
 Los investigadores manipulan especies bacterianas en el intestino utilizando la dieta
Los investigadores manipulan especies bacterianas en el intestino utilizando la dieta
 Complicaciones graves de COVID-19 relacionadas con la ruptura de la barrera intestinal
Complicaciones graves de COVID-19 relacionadas con la ruptura de la barrera intestinal
 ¿Las etiquetas de los productos comerciales de kéfir informan correctamente los niveles microbianos?
El microbioma intestinal es una parte esencial del organismo humano, como ha quedado muy claro a partir de muchas investigaciones realizadas durante las últimas décadas. La Asociación Científica Inter
¿Las etiquetas de los productos comerciales de kéfir informan correctamente los niveles microbianos?
El microbioma intestinal es una parte esencial del organismo humano, como ha quedado muy claro a partir de muchas investigaciones realizadas durante las últimas décadas. La Asociación Científica Inter
 El enjuague bucal afecta los efectos del ejercicio
Los científicos saben desde hace mucho tiempo que la presión arterial desciende después del ejercicio, pero el mecanismo no se ha dilucidado por completo. Un nuevo estudio muestra que las bacterias en
El enjuague bucal afecta los efectos del ejercicio
Los científicos saben desde hace mucho tiempo que la presión arterial desciende después del ejercicio, pero el mecanismo no se ha dilucidado por completo. Un nuevo estudio muestra que las bacterias en
 Los coronavirus humanos necesitan materiales orgánicos para transferirse de manera eficiente entre superficies
El síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2) ha infectado rápidamente a más de 191 millones de personas en todo el mundo desde su aparición a fines de diciembre de 2019. El virus c
Los coronavirus humanos necesitan materiales orgánicos para transferirse de manera eficiente entre superficies
El síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2) ha infectado rápidamente a más de 191 millones de personas en todo el mundo desde su aparición a fines de diciembre de 2019. El virus c